OPTimized inducible KnockOut (OPTiKO)
This is an abridged and updated version of our original protocol paper, which can be downloaded here: OPTiKO
Some plasmids can be obtained from Addgene (https://www.addgene.org/Ludovic_Vallier/); contact us for the Zinc Finger Nucleases plasmids
Some plasmids can be obtained from Addgene (https://www.addgene.org/Ludovic_Vallier/); contact us for the Zinc Finger Nucleases plasmids
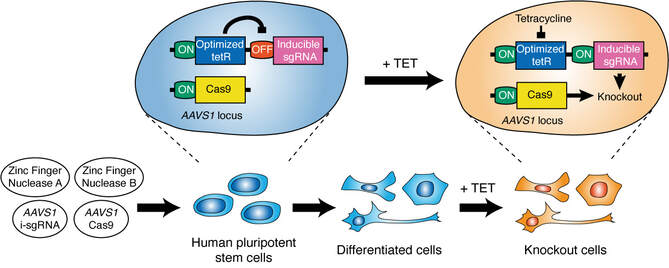
Figure 1. The optimized inducible knockout (OPTiKO) system. Schematic of the generation of OPTiKO human pluripotent stem cells (hPSCs) and of the resulting genetic circuit which allows tetracycline (TET)-dependent induction of gene knockout both in hPSCs and hPSC-derived cells
The following protocol describes all of the procedures required for the generation and validation of OPTiKO hPSCs. First, the sgRNA specific to the gene of interest is designed and cloned as a double-strand oligonucleotide into pAAV-Puro_siKO (Fig. 2). Then, hPSCs are nucleofected with the AAVS1 ZFN plasmids and targeting vectors, and dual-gene targeted lines are selected using puromycin and neomycin (Fig.3). Clonal hPSC lines are isolated and characterized by genomic PCR to identify correctly targeted clones (Fig.4). Finally, the resulting OPTiKO hPSCs are validated to induce efficient gene knockout by confirming loss of the protein product upon tetracycline treatment (Fig.5). The whole procedure can be completed in 4-9 weeks depending on the experimental design and expertise of the investigator and allows generation of OPTiKO hPSCs with >90% efficiency. The protocol shares similarities with a related method to generate inducible knockdown hPSCs, which can serve as additional reference [16, 24].
Materials
Prepare all solutions using analytical grade chemicals, and DNase- and RNase-free ultrapure deionized water (dH2O). Store all reagents at room temperature, unless indicated otherwise below or in the supplier’s instructions. Hazardous reagents must be handled with care while wearing appropriate personal protective equipment and in accordance with local safety regulations. Waste materials are to be disposed according to the relevant regulations. Standard equipment used in molecular biology and cell culture research is required for this protocol (e.g., micropipettes, a tabletop microcentrifuge, and a serological pipette controller). Certain materials are required at multiple stages of the protocol.
Molecular cloning
Gene targeting
Genotyping and validation
Prepare all solutions using analytical grade chemicals, and DNase- and RNase-free ultrapure deionized water (dH2O). Store all reagents at room temperature, unless indicated otherwise below or in the supplier’s instructions. Hazardous reagents must be handled with care while wearing appropriate personal protective equipment and in accordance with local safety regulations. Waste materials are to be disposed according to the relevant regulations. Standard equipment used in molecular biology and cell culture research is required for this protocol (e.g., micropipettes, a tabletop microcentrifuge, and a serological pipette controller). Certain materials are required at multiple stages of the protocol.
Molecular cloning
- pAAV-Puro_siKO plasmid (Addgene #86696).
- Oligonucleotides for sgRNA target (custom).
- AarI restriction enzyme (2 U/µL; ThermoFisher Scientific).
- DNA loading dye (6X): 60% glycerol, 10 mM Tris-HCl (pH 7.6), 60 mM EDTA, 0.03% bromophenol blue, 0.03% xylene cyanol.
- TAE electrophoresis buffer (50X): 2 M Tris base, 1 M acetic acid, 50 mM EDTA disodium salt.
- Agarose I, molecular biology grade
- Ethidium bromide: 10 mg/ml stock solution in dH2O.
- Agarose gel in TAE: dissolve the required amount of agarose powder into 100 mL of 1X TAE in a glass bottle. Place a cap on the bottle but leave loose. Incubate at room temperature for 15’ to pre-dissolve. Microwave for about 1-2’ or until all of the powder is fully dissolved, but do not let the solution boil. Allow the solution to cool at room temperature for 5-10’ (the temperature of the solution should not go below 65 °C to prevent premature gelling), and add ethidium bromide to a final concentration of 0.5 µg/mL. After mixing pour the solution into a gel casting tray equipped with the appropriate combs and let the gel set for 30 minutes before use.
- DNA electrophoresis apparatus.
- DNA molecular weight ladder.
- UV transilluminator.
- QIAEX II Gel Extraction Kit (QIAGEN).
- Oligo annealing buffer (10X): 100 mM Tris-HCl (pH 8), 10 mM EDTA, 1 M NaCl.
- Thermocycler with heated lid.
- T4 DNA ligase (400 U/µL; New England Biolabs).
- α-select Gold Efficiency E. Coli (≥10E09 cfu/μg; Bioline)
- Heated water bath.
- Humidified bacterial incubator.
- S.O.C. medium: 2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose. Adjust to pH 7.0 and sterilize by passing through a 0.22 µm filter.
- Luria Bertani (LB) broth: 1 % tryptone, 0.5 % yeast extract, 171 mM NaCl. Adjust to pH 7.0 and sterilize by autoclaving.
- Ampicillin: 100 mg/ml stock solution in dH2O, store at -20 ºC.
- LB-agar ampicillin bacterial culture petri dishes: dissolve 20 g of agar powder in 1 L of LB broth and mix well. Autoclave on a liquid cycle and let to cool until it is warm enough to touch (approximately 50 °C). Add ampicillin to a concentration of 100 µM and swirl to mix (do not shake as this will create bubbles). Pour into petri dishes to completely cover the bottom surface. Allow the plates to set for 2 h at room temperature, and store sealed plates at 4 °C for up to 3 months.
- Bacterial culture orbital shaker.
- QIAprep Spin Miniprep Kit (QIAGEN).
- siKO_fw primer: 5’-CGAACGCTGACGTCATCAACC-3’
- Glycerol for molecular biology (>99%).
- QIAfilter Plasmid Midi Kit (QIAGEN).
Gene targeting
- Human pluripotent stem cells (in house or from various commercial suppliers).
- 100 mm tissue culture petri dishes.
- TeSR-E8 medium (STEMCELL Technologies).
- 250 µg/mL Vitronectin XF (STEMCELL Technologies).
- UltraPure 0.5 M EDTA pH 8.0 (Life Technologies).
- Humidified tissue culture incubator with CO2 supply.
- Biosafety level 2 laminar air flow tissue culture hood.
- Y-27632 dihydrochloride: 10 mM solution in DMSO. Prepare single-use aliquots and store at -20 °C for up to 6 months.
- DPBS no calcium and no magnesium.
- StemPro Accutase Cell Dissociation Reagent (ThermoFisher Scientific).
- P3 Primary Cell 4D-Nucleofector X Kit L (Lonza).
- Trypan blue: 0.4% w/vol solution in dH2O.
- Hemocytometer.
- pZFN_AAVS1-R-KKR plasmid [contact us, unfortunately these plasmids are not yet available on Addgene].
- pZFN_AAVS1-L-ELD plasmid [contact us, unfortunately these plasmids are not yet available on Addgene]/
- pAAV-Neo_CAG-Cas9 plasmid (Addgene #86698).
- pAAV-Puro_siKO-sgRNA plasmid (custom).
- AAVS1-CAGGS-EGFP (Addgene #22212).
- 4D-Nucleofector Core Unit and X Unit (Lonza)
- Puromycin dihydrochloride: 10 mg/ml solution in dH2O. Sterilize by passing through a 0.22 µm filter, and store at -20 °C for up to 1 year.
- Geneticin (G418 sulfate): 200 mg/ml solution in dH2O. Sterilize by passing through a 0.22 µm filter, and store at -20 °C for up to 1 year.
Genotyping and validation
- 24 well tissue culture petri dishes.
- 10.000 U/ml penicillin-streptomycin.
- Wizard SV Genomic DNA Purification Kit (Promega)
- LongAmp Taq DNA Polymerase (2.5 U/µL; New England Biolabs)
- dNTP mix: 10 mM dATP, 10 mM dCTP, 10 mM dGTP, 10 mM dTTP.
- Dimethyl sulfoxide (DMSO), PCR grade.
- Genotyping primers (see Table 1 for full list and sequences): 5 µM stock solutions in dH2O.
- Tetracycline hydrochloride (cell-culture grade; Sigma-Aldrich): to prepare a stock 10 mg/ml solution dissolve 50 mg of tetracycline hydrochloride in 5 ml of dH2O. The resulting solution should have a mild yellow-orange color. Filter-sterilize using a 0.22 µm filter, and prepare single-use 5 or 10 µl aliquots. Store at -80 °C for up to 6 months. Prepare and store this reagent protected from direct illumination.
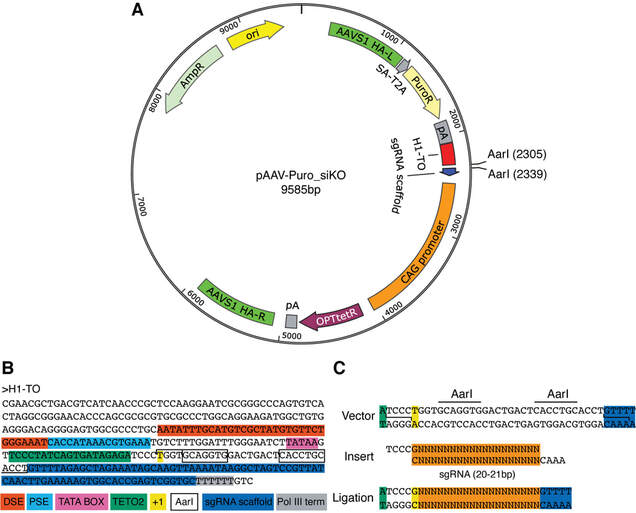
Figure 2. Cloning of sgRNAs into the OPTiKO plasmid. (a) Map of the AAVS1 targeting vector carrying the inducible sgRNA: the transgene is integrated following homologous directed repair (HDR), and employs a gene-trap approach to drive the drug resistance gene through the endogenous promoter of the AAVS1 locus (Fig. 4). HA-L/R left/right homology arms, SA splice acceptor, T2A self-splicing T2A sequence, PuroR puromycin resistance cDNA, pA polyadenylation sequence, H1-TO tetracycline-inducible H1 RNA Polymerase III promoter containing a tetO2 sequence, CAG CMV early enhancer, chicken β-actin, and rabbit β-globin hybrid constitu- tive promoter, OPTtetR codon-optimized tetracycline-responsive repressor protein cDNA, AmpR ampicillin resistance, ori high-copy origin of replication. (b) Nucleotide sequence of the tetracycline-inducible H1-TO RNA Polymerase III promoter. Key features are color coded. The restriction enzyme sites used for sgRNA cloning are shown in boxes (panel c). DSE distal sequence element, PSE proximal sequence element, TETO2 tet operator; +1 start position of RNA transcription, Pol III term poly(T) sequence inducing transcription termination. (c) Schematics of the sgRNA cloning procedure.
Generation of inducible sgRNA targeting vector
- Identify a 20 bp sgRNA target sequence against the gene of interest by taking advantage of available sgRNA design tools (see Note 1). Testing of the candidate sgRNAs efficiency is optional but is strongly recommended (see Note 2). If the sgRNA sequence does not start with a guanine (“G”) or an adenosine (“A”), add a “G” to the 5’ end (see Note 3).
- Design the “top” oligonucleotide by adding 5’-TCCC-3’ to the 5’ end of the sgRNA target sequence (without PAM), and the “bottom” oligonucleotide by adding 5’-AAAC-3’ to the 5’ end of the reverse complement of the sgRNA target sequence (without PAM). Annealing of these oligonucleotides will create a short double-strand DNA sequence with 5’ overhangs suitable for subsequent directional cloning (Fig. 2c). Order both oligonucleotides as desalted purified products from a preferred vendor, and resuspend at a concentration of 200 µM in dH2O.
- Prepare the backbone plasmid by digesting 5 µg of pAAV-Puro_siKO in a 100 µL reaction containing 5 µL (10 U) of AarI, 2 µL (0.5 µM) of the provided oligonucleotide, and 10 µL (1X) of the provided 10X AarI digestion buffer (remaining volume dH2O). Incubate at 37 ºC overnight (16 h; see Note 4). AarI is a type II restriction enzyme that cuts outside of its two recognition sites to create non-complementary 3’ overhangs suitable for seamless directional cloning of the sgRNA target sequence in front of the sgRNA scaffold (Fig. 2a-c).
- Add 20 µL (1X) of the 6X DNA gel loading dye, and perform standard DNA electrophoresis of the digestion product using a 1% agarose-TAE gel. Include a DNA ladder as molecular weight control. Visualize the DNA using a UV transilluminator, excise the 9551 bp band with a clean scalpel (see Note 5), and gel-extract the DNA using QIAEX II Gel Extraction Kit following the manufacturer’s instructions. Quantify the linearized plasmid, and adjust the concentration to 50 ng/µL. Keep on ice for same day use, or store in single-thaw aliquots at -20 °C for at least 6 months.
- Prepare the insert by annealing the top and bottom oligonucleotides from step 2 in a 20 µL reaction containing 5 µL (50 µM) of each oligo and 2 µL (1X) of 10X oligo annealing buffer (remaining volume dH2O). Incubate in a thermocycler for 5’ at 95 °C, followed by slow descent to 4 °C by -0.1 °C/s, then dilute 1:500 in 1X oligo annealing buffer. Keep on ice for same day use (do not store).
- Assemble a 10 µL ligation reaction with 4 µL of the diluted annealed oligo, 1 µL (50 ng) of the linearized plasmid from step 4, 1 µL (5 U) of T4 DNA Ligase, and 1 µL (1X) of the provided 10X T4 DNA Ligase Buffer (remaining volume dH2O). Incubate at room temperature for 2 h.
- Transform 2 µL of the ligation product into 25 µL of α-select E. Coli according to manufacturer’s instructions using a heated water bath for heat-shock. Recover the transformed bacteria in 250 µL of S.O.C. media for 30’ at 37 °C, then plate all of the culture onto a LB-agar petri dish containing 100 µg/ml of ampicillin. Incubate overnight (16 h) at 37 °C in a humidified incubator.
- Pick individual bacterial colonies using a sterile tip and inoculate them into 4 ml of LB broth with 100 µg/ml of ampicillin (see Note 6). Incubate in a bacterial culture orbital shaker at 37 °C overnight (16 hours) while shaking at 225 rpm
- Isolate plasmids from bacterial cultures using the QIAprep Spin Miniprep Kit following manufacturer’s instructions. Save 200 µl of each culture and store in the fridge.
- Confirm the presence of the desired sgRNA and the lack of mutations by performing Sanger DNA sequencing using the siKO_fw primer. Upon successful result, freshly inoculate the bacterial clone by diluting the previously saved liquid culture 1:1000 in 50 mL of LB broth with 100 µg/ml of ampicillin. Incubate ina bacterial culture orbital shaker at 37 °C overnight (16 h) while shaking at 225 rpm.
- Prepare a glycerol stock to be stored at -80 °C for long-term backup by mixing 200 µl of sterile autoclaved 50% glycerol in dH2O with 200 µL of the bacterial culture. Isolate the plasmid from the remaining culture using the QIAfilter Midiprep Kit according to the manufacturer’s instructions. Resuspend the resulting pAAV-Puro_siKO-sgRNA targeting vector in dH2O at a concentration of 1 µg/mL and store at -20 °C.
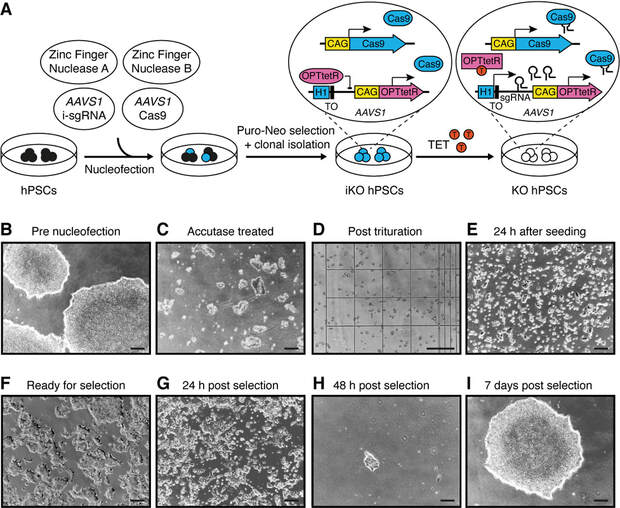
Figure 3. Generation of OPTiKO hPSCs by gene targeting. (a) Schematic of the gene targeting procedure. The resulting OPTiKO transgenic alleles and their functionality in the presence or absence of tetracycline (TET) are shown. ZFN zinc finger nuclease, i-sgRNA inducible sgRNA, iKO inducible knockout, KO knockout. (b–i) Representative phase-contrast images of hPSCs at the indicated stages of the gene targeting procedure. Scale bars: 250 μM.
Gene targeting of inducible CRISPR/Cas9 platform in hPSCs
- Grow hPSCs in mTeSR E8 medium and vitronectin-coated culture dishes according to the manufacturer’s instructions in a 37°C incubator with 5% CO2 and 95% humidity. Passage cells every 3 to 5 days using 0.5 mM EDTA to facilitate mechanical dissociation, and seed them as small clumps of 5-20 cells with sub-cultivation ratio of 1:3 to 1:10 (see Note 7). hPSCs to be used for nucleofection should be within their exponential growth phase (50-70% confluency; Fig. 3b), and 2x106 cells are required for each nucleofection. Positive and negative nucleofection controls are strongly recommended for first time users or when troubleshooting (see Note 8). Volumes are given for cells cultured in 100 mm dishes.
- 16-24 h before nucleofection feed hPSCs with 10 mL of fresh TeSR-E8 media without antibiotics and supplemented with 10 µM Y-27632 (ROCK inhibitor; see Note 9).
- Before beginning the nucleofection procedure, prepare two vitronectin-coated 100 mm dishes for each nucleofection, aspirate the coating solution, add 10 mL of TeSR-E8 medium supplemented with 10 µM Y-27632, and place in the 37°C incubator to pre-warm and equilibrate. Thaw all the plasmids and adjust their concentration to 1 µg/ml. Add the supplement to the P3 nucleofection solution from the 4D-Nucleofector kit, and let acclimatize at room temperature.
- Begin cell collection by aspirating hPSC culture medium, rinsing the cells with 10 mL DPBS, and adding 5 ml of Accutase. Incubate for 3 to 5 min at 37 °C in the incubator until colonies can be lifted by gently tapping on the side of the dish (Fig. 3c). Add 10 mL of TeSR-E8 medium, and mechanically triturate the colonies into clumps of 3-4 cells using a 5 mL serological pipette (Fig. 3d).
- Perform a live cell count using a hemocytometer after diluting an aliquot of the cells 1:2 with trypan blue (see Note 10). Aliquot 2x106 live cells in a separate conical tube for each nucleofection, and pellet the cell suspension by spinning for 3’ at 115 g at room temperature. Remove the supernatant as completely as possible.
- Prepare the nucleofection mix in a sterile tube by adding 2 µL (2 µg) of pAAV-Puro_siKO-sgRNA, 2 µL (2 µg) of pAAV-Neo_CAG-Cas9, 4 µL (4 µg) of pZFN_AAVS1-R-KKR, 4 µL (4 µg) of pZFN_AAVS1-L-ELD, and finally 100 µL of P3 solution. Using a 1000 µL pipette tip transfer all of the mix to the tube containing the cell pellet, and resuspend the cells very gently by pipetting 3-5 times (see Note 11).
- Rapidly transfer the nucleofection mix to the provided nucleofection cuvette (see Note 12), and pulse the cells by operating the program “CA-137” (see Note 13). Let the cells recover at room temperature for 5’.
- Add 500 µL of TeSR-E8 medium supplemented with 10 µM Y-27632 to the bottom of the cuvette, and using the provided suction pipette very gently transfer half of the cell suspension to each of the two pre-warmed 100 mm dishes from step 3. Distribute drop by drop over the plate surface, and gently shake the plate back and forth then left and right 3-5 times to promote even plating. Incubate overnight in the 37 °C incubator (see Note 14).
- On the following day, visually confirm efficient cell attachment (Fig. 3e), and replace the culture media with fresh TeSR-E8 medium. Subsequently, perform daily media changes. After 3 days from nucleofection, or when cells reach 50-70% confluence (Fig. 3f), begin dual drug selection by adding 0.5 µg/ml of puromycin and 25 µg/ml geneticin to the culture media (see Note 15). For the first 48 h of selection, further supplement the media with 10 µM Y-27632 (see Note 9). Selection should be complete within 48-72 h (Fig. 3g-h; see Notes 8 and 16), after which the drugs can be withdrawn. Individual colonies should reach an appropriate size for passaging (1-2 mm; Fig. 3i) by 7-10 days after nucleofection (see Note 17).
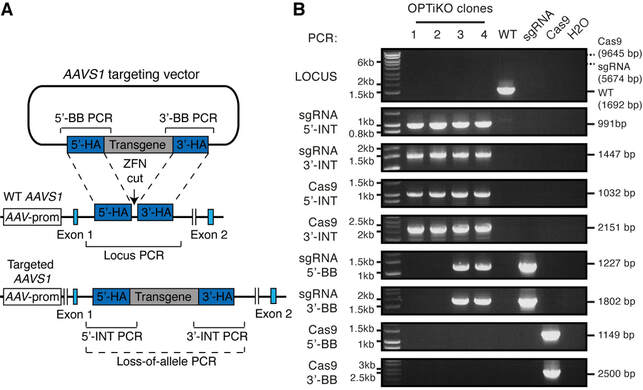
Figure 4. Genotyping of OPTiKO hPSCs. (a) Schematic of the AAVS1 gene targeting event that generates the OPTiKO transgenic alleles through homologous recombination of the donor plasmids (features are not in scale). AAV prom: endogenous promoter of the AAVS1 locus (PPP1R12C gene), which drives the gene-trap drug resistance genes. The genotyping strategies used to identify correctly targeted hPSCs are shown. Locus PCR PCR product of wild-type AAVS1 locus (indicating a non-targeted allele); Loss-of-allele: potential PCR amplification that fails onto the targeted allele due to large size and high GC-content; 5'/3'-INT PCR: PCR product of transgene 5'/3'-end integration region (indicative of expected transgene targeting); 5'/3'-BB PCR: PCR product of vector backbone 5'/3'-end (indicative of nonspecific off- target plasmid integration). (b) Representative example of genotyping results from 4 OPTiKO clonal sublines. For all clones the AAVS1 locus is correctly targeted with both transgenes, while clones 3 and 4 also carry randomly integrated copies of the targeting plasmid (Table 2). All clones could be used for further experiments. The predicted size of each PCR amplicon is indicated (Table 1), and a molecular weight control is shown on the left. WTcontrol PCR from wild-type hPSCs, sgRNA control PCR from pAAV-Puro_siKO plasmid, Cas9 control PCR from pAAV-Neo_CAG-Cas9 plasmid, H2O no template control.
3.3 Genotyping of OPTiKO hPSCs
- Mechanically pick an individual colony by using a micropipette equipped with a sterile tip. Use a microscope to facilitate visualization during the procedure. Transfer each colony to a single well of the 24 well tissue culture petri dish, then gently pipette 5-10 times to triturate. Add penicillin-streptomycin to the culture media to minimize the risk of bacterial contamination, and further supplement it with 10 µM Y-27632 to promote hPSC survival. Repeat the procedure for 8-12 colonies (see Note 18).
- Once they reach 50-70% confluency, split each clonal line into two wells of a 24-well plate: one with approximately 1/3 of the cells and the second with the remaining 2/3. Cells in the first well will be grown, while the second well will be used for genotyping.
- When the cells prepared for genotyping reach >50% confluency, extract genomic DNA using the Wizard Genomic DNA Purification Kit according to the manufacturer’s instructions. Adjust the DNA concentration to 25-50 ng/µL.
- Individually assemble the genotyping polymerase chain reactions (PCR) detailed in Table 1 using LongAmp Taq DNA Polymerase (see Note 19). For each reaction, prepare a 10 µL mix containing: 100 ng of genomic DNA, 0.3 µL (300 µM) dNTP mix, 0.5 µL (250 nM) forward primer, 0.5 µL (250 nM) reverse primer, 0.2 µL (2%) DMSO, and 0.4 µL (10 U) LongAmp Taq Polymerase (remaining volume dH2O). Inclusion of positive and negative controls is strongly recommended (see Note 20).
- Perform the PCR in a thermocycler according to the following program (lid heated at 95 °C): (1) 94 °C for 5’; (2) 94 °C for 15’’; (3) annealing temperature (see Table 1) for 30’’; (4) 65 °C for extension time (see Table 1); (5) repeat steps 2 to 4 for a total of 35 cycles; (6) 65 °C for 3’; (7) hold at 10 °C.
- Perform DNA gel electrophoresis for half of each PCR reaction with a 1% agarose-TAE gel, and visualize the results with a UV transilluminator (Fig. 4b).
- Determine the genotype of OPTiKO clones by referring to Table 2 (see Note 19). Only clones showing dual targeting of both the Cas9 and sgRNA transgenes are OPTiKO hPSCs that should be kept for further experiments.
- Identify the clones of interest from the second 24 well plate from step 1. Once they are ready to be passaged split them for expansion, banking, and experimental analyses.
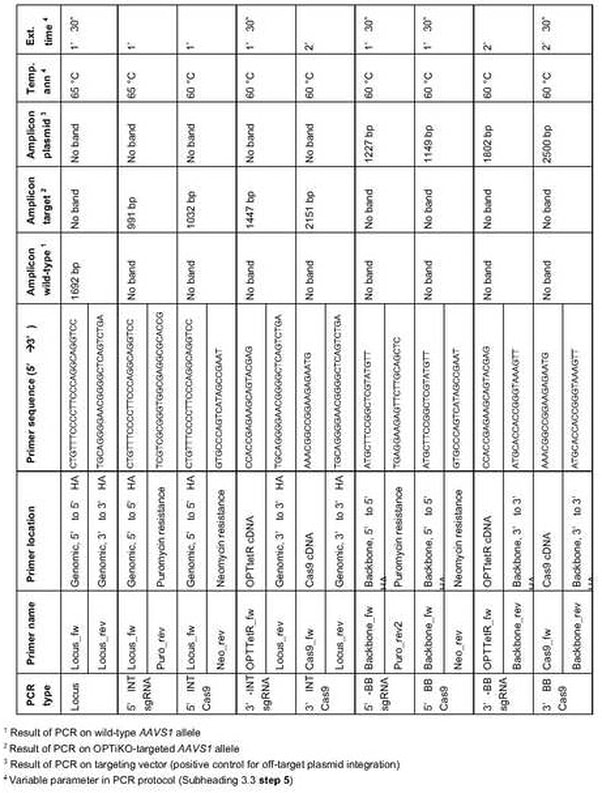
Table 1 – Primers for genotyping of OPTiKO clonal lines.
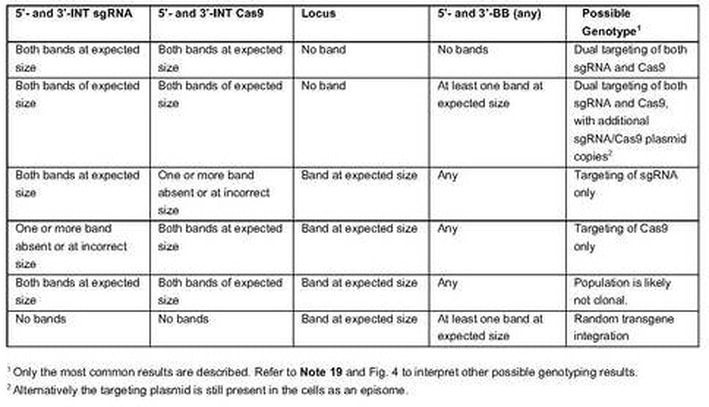
Table 2. Inferring the genotype of OPTiKO clonal lines from PCR results.
3.4 Validation of OPTiKO hPSCs
- Knockout can be induced in hPSCs or hPSC-derived cells by adding the drug tetracycline to the culture media at a concentration of 1 µg/ml (see Notes 21 and 22). The duration of tetracycline treatment required to induce knockout in the majority of the cells will depend on the efficiency of the sgRNA and on the cell type (Fig. 5), but ought to be in the range of 2-10 days (see Note 23).
- Validation of gene knockout should be performed by means of appropriate techniques described in detail elsewhere (see Notes 24 and 25). The quantification of CRISPR/Cas9-induced indels on the genomic locus can be efficiently assessed using the Surveyor or T7 endonuclease 1 assay (Fig. 5a), by DNA Sanger sequencing, and/or by next-generation sequencing [17, 25] (see Note 26). Mutations introducing premature stop codons will often result in decreased transcript levels due to nonsense-mediated decay, which can be easily measured by quantitative reverse transcription PCR (RT-qPCR) [26]. Finally and most importantly, loss of the protein product can be validated by flow cytometry (Fig. 5b), immunocytochemistry (Fig. 5c-e), and/or Western blot [16, 27–30] (see Note 27).
- Validated OPTiKO hPSCs may now be used to investigate the biological question of interest (Fig. 5f; see Note 28)
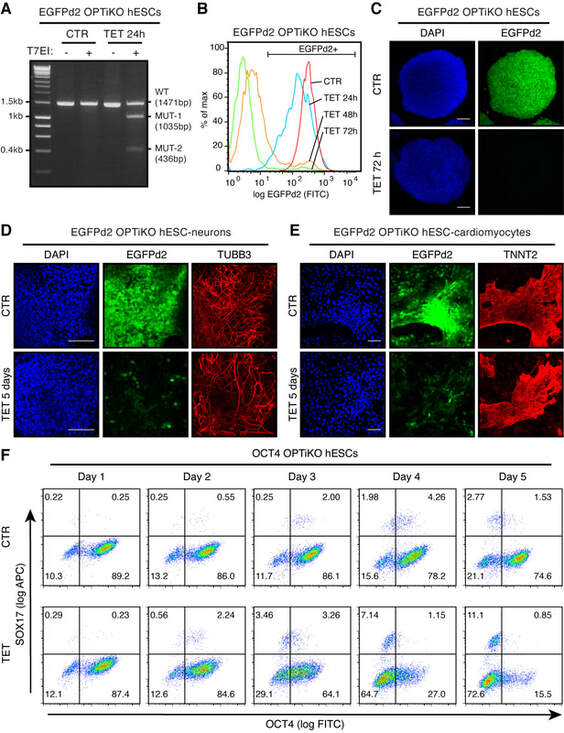
Figure 5. Performance of OPTiKO in hPSCs and hPSC-derived cells. (a) T7 endonuclease 1 (T7E1) assay in EGFPd2 OPTiKO hESCs [16]. hESCs constitutively expressing a destabilized EGFP transgene (homozygous ROSA26 CAG-EGFPd2 hESCs) were retargeted with the OPTiKO plasmids to drive an inducible sgRNA against EGFP. Cells were analyzed in the absence (CTR) or presence of tetracycline (TET) for 24 h. A 1.47 Kb fragment around the sgRNA cut site was amplified by PCR and analyzed by T7E1 assay. The presence of indels in tetracycline-treated cells led to T7E1-mediated cleavage of the genomic fragment into the indicated products (MUT-1 and MUT-2). (b) Quantification of EGFPd2 by flow cytometry in EGFPd2 OPTiKO hESCs. >95% knockout could be achieved following 72 h of induction. (c–e) Analysis by immunocytochemis- try of EGFP2d OPTiKO hESCs (c), hESC-derived neurons (d), and hESC-derived cardiomyocytes (e). EGFPd2 fluorescence is shown in green, while for (d) and (e) lineage-specific markers are shown in red. DAPI: nuclear staining. Scale bars: 100 μM. Widespread loss of EGFPd2 expression could be achieved in all lineages. (f) Analysis by flow cytometry of OCT4 OPTiKO hESCs [17]. Cells were analyzed for expression of OCT4 and of the endoderm marker SOX17. The result demonstrates that loss of OCT4 expression led to endoderm differentiation in a subpopulation of hESCs.
Notes
- sgRNA design should consider several aspects including the cut site location (conserved and constitutive exons located towards the start of the coding sequence and/or encoding for crucial protein domains are to be preferred), and the predicted activity and specificity of the sgRNA [31, 32]. Publicly available resources for sgRNA design are numerous and reviewed elsewhere [33]. We recommend the online tools CRISPR design (http://crispr.mit.edu) or GuideScan [34], or the standalone software Protospacer Workbench [35].
- Even more so than for conventional CRISPR/Cas9 applications, identification of an efficient sgRNA is the most critical factor in the success of OPTiKO experiments [16, 17]. The sgRNA will be expressed from a single transgenic copy, and will therefore be in limiting amounts. As a result, efficient gene editing will be achieved only with a sgRNA highly effective in inducing double-strand DNA breaks (see Note 23). It is therefore highly recommended to pre-screen 3 to 5 sgRNAs using an appropriate method, such as the inexpensive T7 endonuclease I (T7EI) assay [36], and to select the sgRNA showing the highest activity.
- Distinctly from the U6 promoter (which prefers transcription of sgRNAs starting with guanine), either a guanine or an adenosine may support efficient and precise transcription from the H1 Pol III promoter [37, 38]. Addition of a guanine at the start of sgRNAs not satisfying this criterion does not substantially influence gene editing efficiency.
- AarI requires binding to two copies of its recognition sequence for cleavage (the second copy is provided by the oligonucleotide to be included in the digestion reaction), and is a slow cutter that requires extended incubation for complete plasmid digestion.
- Occasional AarI star activity after overnight digestion can lead to the presence non-specific products smaller in size compared to the expected linearized plasmid. Provided that the linearized plasmid is the predominant digestion product and it can be clearly identified and distinguished after an appropriate electrophoretic run, this will not interfere with the efficiency of subsequent ligation. If this is not the case, however, the digestion time should be shortened to 4-8 h.
- We recommend screening 4 to 8 bacterial clones. If the procedure was successful, the expected recombinant DNA should be found it more than 90% of the clones. Should the efficiency prove significantly lower, colony PCR can be used as a rapid method to screen several clones [16]. For this, use the siKO_fw primer and the bottom sgRNA oligonucleotide as reverse primer in order to detect an approximately 250 bp product in the clones carrying the sgRNA. Additionally, correct recombinant clones can be screened by diagnostic digestion with AarI, as this will be unable to digest the desired recombinant DNA (provided no AarI site is found within the sgRNA sequence).
- The culture conditions recommended in this protocol rely on commercially available and commonly used reagents for feeder- and xeno-free hPSC culture. Nevertheless, OPTiKO hPSCs have been successfully derived from cells maintained in various culture conditions including gelatin-fibronectin coating with an in house chemically defined media, matrigel coating with mTeSR1, and laminin-521 coating with E8 [16, 17].
- A positive control can be performed by using an AAVS1 targeting vector expressing a constitutive EGFP transgene (AAV-CAGGS-EGFP) instead of the OPTiKO plasmids. Cells can be visually monitored for EGFP expression the day following nucleofection to confirm efficient plasmid delivery, and the nucleofected cells can be subjected to drug selection to confirm efficient gene targeting of the AAVS1 locus. Note that this targeting vector confers resistance only to puromycin, so geneticin should be omitted. A negative control nucleofection without any plasmid can be performed to monitor the efficiency of drug selection: the nucleofected cells should be completely killed within 48-72 h of drug selection. Finally, an additional negative control nucleofection only with the OPTiKO plasmids but omitting the AAVS1 zinc finger nucleases can be performed to confirm the low rate of transgene integration outside of the AAVS1 locus: no or very few colonies should survive drug selection.
- The use of antibiotics is optional at all other stages of the procedure but must be avoided the day before and the day after the nucleofection, as they are toxic in cells with increased cell membrane permeability. The addition of ROCK inhibitor before and after nucleofection increases hPSC survival by inhibiting apoptosis [39].
- Viability should be greater than 90%. If this is significantly lower, increase the duration of Accutase treatment up to 7’ to reduce the amount of mechanical trituration to the minimum required to obtain 3-4 cell clumps.
- Use of a pipette tip with a large orifice is key to limit shear stress on nucleofected cells. Pipetting should be very gentle and minimized as much as possible. In particular, cells post-nucleofection are incredibly fragile and should be mixed only a couple of times, preferably using a large bore pipette tip.
- Ensure that no air bubbles are trapped at the bottom of the cuvette. If so, a sharp tap should be sufficient to dislodge them.
- The combination of the recommended nucleofection solution and nucleofection program has been optimized for H9 hESCs and proved to work efficiently also in hiPSCs (40-60% nucleofection efficiency). Nevertheless, this might require some degree of optimization for certain hPSC lines. Consult the nucleofector manufacturer’s instructions for alternative nucleofection solutions and programs suitable for hPSCs.
- An alternative to the recommended feeder-free procedure is plating the nucleofected cells onto feeder layers of mitotically inactivated MEF cells [16]. While more expensive, laborious, and not xenofree, this approach may increase gene editing efficiency of certain hPSC lines due to improved survival both after nucleofection and during drug selection. In this case, utilize DR4 MEF (Applied StemCell) that are genetically engineered to contain resistance to puromycin and neomycin, and top up the feeder layer as needed during drug selection (some cell death of the MEF feeder will be observed despite their drug resistance).
- The recommended drug doses have been optimized for H9 hESCs, and proved to work efficiently also for hiPSCs. Nevertheless, based on the results of the positive and negative controls described in Note 8, optimization might be required for certain hPSC lines. In this case, perform a kill curve experiment to identify the minimal dose required to completely select wild-type hPSCs within 48-72 h of drug selection (range: 0.25-2 µg/ml of puromycin and 12.5-200 µg/ml geneticin).
- During the first 48 h of selection it can be beneficial to increase the media volume to 20 mL and to perform media changes every 12 h in order to rapidly remove dead cells, thus reducing the stress they impose on the surviving hPSCs.
- 5-50 individual hPSC colonies should be obtained in each of the two 100 mm dish. An individual colony is expected to result from clonal expansion of a single cell that experienced the rare gene editing event in which both AAVS1 alleles have been targeted with the two different transgenes. Therefore, provided that such a pseudo-clonal colony is not in close proximity to any other, this will be considered and referred to as “clonal”. Should a more stringent way to generate clonal lines be desirable, hPSCs should be plated as single cells into 96 or 384 well plates and grown as bona fide clones.
- Given the high efficiency of the gene targeting procedure, the proportion of correctly targeted hPSCs is expected to be greater than 90% [16]. As a matter of fact, while clonal isolation is recommended in order to obtain an isogenic population, this may be entirely bypassed in experimental situations where this is not an important requirement (such as for analyses that will be performed at the single cell level). In such case, simply passage the whole 100 mm dishes into new plates and proceed to validation.
- The recommended genotyping strategies are illustrated in Fig. 4a. The 5’- and 3’-integration (INT) PCRs are designed to verify site-specific integration of the transgenes. Targeting of both alleles of the AAVS1 locus is further confirmed by loss-of-allele PCR for the wild type locus (this PCR fails following successful transgene integration due to the large size of the amplicon and the high GC content of the CAG promoter). Finally, 5’- and 3’-backbone (BB) PCRs allow to determine if the targeting plasmids have been integrated in a random genomic region. Note that the BB PCRs are optional, as the presence of additional transgenic copies does not interfere with the functionality of the method. Nevertheless, selection of clones with exactly one copy of each transgene can be advantageous in certain experimental settings, for instance when comparing the efficiency of different sgRNAs [17].
- Recommended controls are wild-type genomic DNA, 100 ng; pAAV-Neo_CAG-Cas9 (Cas9 targeting plasmid), 1 ng; pAAV-Puro_siKO-sgRNA (sgRNA targeting plasmid), 1 ng; and no template control, dH2O (Fig. 4b).
- This dose of tetracycline is not toxic to hPSCs and does not interfere with hPSC differentiation into multiple lineages from all the germ layers [16]. Nevertheless, this might require optimization for a specific sensitive cell type (range: 0.05-2 µg/ml). Tetracycline is unstable in aqueous solutions and should be added fresh from single-use freshly-thawed aliquots (see Materials). Media changes should be performed at least every other day since the half-life of tetracycline at 37 °C is of approximately 24 h.
- Given the widespread use of tetracycline as an antibiotic in livestock animals, animal-derived products such as fetal bovine serum (FBS) may be contaminated with tetracycline. When such reagents have to be included as part of the culture media to be used for hPSC maintenance or differentiation, batch testing to confirm lack of detectable tetracycline contamination is strongly recommended. Tetracycline-free FBS is available from commercial suppliers.
- As introduced in Note 2, the speed and efficiency of knockout will be predominantly dictated by the activity of the sgRNA. Should the performance of a given sgRNA be insufficient to appropriately investigate the biological question at hand, an alternative is to build a pAAV-Puro_siKO plasmid containing multiple copies of the sgRNA. This can be achieved by a one-step Gibson assembly reaction using PCR-amplified inducible sgRNA expression cassettes [16]. Of note, this method can also be applied to express multiple distinct sgRNAs against the same gene, or multiple sgRNAs against different genes.
- For most sgRNAs the level of gene knockout in absence of the inducer tetracycline should be minimal (less than 5%) even after prolonged passaging [16, 17]. However, rare exceptionally potent sgRNAs can induce even substantial premature gene knockout. In such case, it is recommended to repeat the procedure using an alternative targeting vector for sgRNA cloning: pAAV-Puro_siKO-2TO (Addgene #86697). In such plasmid the inducible H1 promoter has an additional tetO2 binding side for the tetR before the TATA box, thus reinforcing transcriptional repression in the absence of tetracycline [16, 40]. Note that this modification also results in lower sgRNA expression levels after addition of tetracycline, thus making it a valuable option only in case of exceptionally strong sgRNAs.
- The kinetics by which acquisition of knockout mutations leads to loss of the protein product is heavily dependent on the stability of both the mRNA and the protein. Furthermore, decrease in protein levels will be more marked in dividing cells (which will serially dilute the protein after each cell cycle) compared to non-proliferative cells. Therefore, the timing required for efficient knockout ought to be determined for each gene and cell type to be studied.
- The same methods should also be applied to determine the degree of possible CRISPR/Cas9 off-targets effects based on in silico predictions. Indeed, while these ought to be minimized by careful design of the sgRNA (see Note 1), the method relies on wild type Streptococcus Pyogenes Cas9 protein, which is known to suffer from some degree of off-target activity [41, 42].
- The OPTiKO method can be also applied to introduce indels in non-coding regions of the genome, which represent an important proportion of disease-associated sequences [1, 2]. In this case, validation will primarily focus on determining the mutations at the DNA level.
- Analysis of OPTiKO cells maintained in parallel and cultured in absence of tetracycline must be always implemented to provide a reference for the experiment. Additionally, when investigating a new cell type and/or biological process we recommend to also analyze cells treated with tetracycline but lacking the specific inducible sgRNA, in order to control for potential non-specific effects due to the drug [43, 44]. For this, OPTiKO cells carrying a scrambled sgRNA are the ideal control, as they also allow to monitor for potential non-specific effects of CRISPR/Cas9 expression. Alternatively, use wild-type cells, or cells expressing the Cas9 transgene. Since clonal isolation of hPSCs can exacerbate biological variability, it is important to analyze multiple OPTiKO clones for the same sgRNA to ensure that the results are reproducible. Finally, analysis of OPTiKO cells carrying separate sgRNAs designed to target the same gene will reinforce the conclusion of any experimental analysis by ruling out the possibility that the observed phenotypes are due to unappreciated off targets of the sgRNA.
References
- Cooper GM, Shendure J (2011) Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12:628–40
- Zhang F, Lupski JR (2015) Non-coding genetic variants in human disease. Hum Mol Genet 24:R102–R110
- Trounson A, DeWitt ND (2016) Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol 17:194–200
- Avior Y, Sagi I, Benvenisty N (2016) Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol 17:170–182
- Murry CE, Keller G (2008) Differentiation of Embryonic Stem Cells to Clinically Relevant Populations: Lessons from Embryonic Development. Cell 132:661–680
- Cong L, Ran FA, Cox D, et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–23
- Mali P, Yang L, Esvelt KM, et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–6
- Jinek M, Chylinski K, Fonfara I, et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–21
- Wright AV, Nuñez JK, Doudna JA (2016) Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164:29–44
- Hsu PD, Lander ES, Zhang F (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157:1262–1278
- González F, Zhu Z, Shi Z-D, et al (2014) An iCRISPR Platform for Rapid, Multiplexable, and Inducible Genome Editing in Human Pluripotent Stem Cells. Cell Stem Cell 15:215–26
- Chen Y, Cao J, Xiong M, et al (2015) Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell 17:233–244
- Mandegar MA, Huebsch N, Frolov EB, et al (2016) CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18:541–53
- Haenebalcke L, Goossens S, Naessens M, et al (2013) Efficient ROSA26-based conditional and/or inducible transgenesis using RMCE-compatible F1 hybrid mouse embryonic stem cells. Stem Cell Rev 9:774–85
- Ordovas L, Boon R, Pistoni M, et al (2015) Efficient recombinase-mediated cassette exchange in hPSCs to study the hepatocyte lineage reveals AAVS1 locus-mediated transgene inhibition. Stem Cell Reports 5:918–931
- Bertero A, Pawlowski M, Ortmann D, et al (2016) Optimized inducible shRNA and CRISPR/Cas9 platforms for in vitro studies of human development using hPSCs. Development 143:4405–18
- Fogarty NME, McCarthy A, Snijders KE, et al (2017) Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550:
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550–3
- Smith JR, Maguire S, Davis L a, et al (2008) Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 26:496–504
- Hockemeyer D, Soldner F, Beard C, et al (2009) Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27:851–7
- DeKelver RC, Choi VM, Moehle E a, et al (2010) Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res 20:1133–42
- Pawlowski M, Ortmann D, Bertero A, et al (2017) Inducible and Deterministic Forward Programming of Human Pluripotent Stem Cells into Neurons, Skeletal Myocytes, and Oligodendrocytes. Stem Cell Reports 8:803–812
- Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
- Bertero A, Yiangou L, Brown S, et al (2018) Conditional manipulation of gene function in human cells with optimized inducible shRNA. Curr Protoc Stem Cell Biol 44:5C.4.1-5C.4.49
- Santos DP, Kiskinis E, Eggan K, et al (2016) Comprehensive Protocols for CRISPR/Cas9-based Gene Editing in Human Pluripotent Stem Cells. Curr Protoc Stem Cell Biol 38:5B.6.1-5B.6.60
- Fan H, Robetorye RS (2010) Real-time quantitative reverse transcriptase polymerase chain reaction. Methods Mol Biol 630:199–213
- Wang L, Gaigalas AK, Yan M (2011) Quantitative Fluorescence Measurements with Multicolor Flow Cytometry. In: Methods in molecular biology (Clifton, N.J.). pp 53–65
- Willingham MC (2010) Fluorescence Labeling of Intracellular Antigens of Attached or Suspended Tissue-Culture Cells. Methods Mol Biol 588:153–164
- Willingham MC (2010) Fluorescence Labeling of Surface Antigens of Attached or Suspended Tissue-Culture Cells. Methods Mol Biol 588:143–151
- Komatsu S (2015) Western Blotting Using PVDF Membranes and Its Downstream Applications. Methods Mol Biol 1312:227–36
- Tsai SQ, Joung JK (2016) Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat Rev Genet 17:300–312
- Mohr SE, Hu Y, Ewen-Campen B, et al (2016) CRISPR guide RNA design for research applications. FEBS J 283:3232–8
- Graham DB, Root DE (2015) Resources for the design of CRISPR gene editing experiments. Genome Biol 16:260
- Perez AR, Pritykin Y, Vidigal JA, et al (2017) GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 35:347–349
- MacPherson CR, Scherf A (2015) Flexible guide-RNA design for CRISPR applications using Protospacer Workbench. Nat Biotechnol 33:805–6
- Bloom K, Ely A, Arbuthnot P (2017) A T7 Endonuclease I Assay to Detect Talen-Mediated Targeted Mutation of HBV cccDNA. Methods Mol Biol 85–95
- Ranganathan V, Wahlin K, Maruotti J, Zack DJ (2014) Expansion of the CRISPR-Cas9 genome targeting space through the use of H1 promoter-expressed guide RNAs. Nat Commun 5:4516
- Ma H, Wu Y, Dang Y, et al (2014) Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Mol Ther 3:e161
- Watanabe K, Ueno M, Kamiya D, et al (2007) A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25:681–6
- Henriksen JR, Løkke C, Hammerø M, et al (2007) Comparison of RNAi efficiency mediated by tetracycline-responsive H1 and U6 promoter variants in mammalian cell lines. Nucleic Acids Res 35:e67
- Slaymaker IM, Gao L, Zetsche B, et al (2015) Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88
- Kleinstiver BP, Pattanayak V, Prew MS, et al (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–5
- Chatzispyrou IA, Held NM, Mouchiroud L, et al (2015) Tetracycline Antibiotics Impair Mitochondrial Function and Its Experimental Use Confounds Research. Cancer Res 75:4446–4449
- Moullan N, Mouchiroud L, Wang X, et al (2015) Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep 10:1681–1691