OPTimized inducible KnockDown (OPTiKD)
This is an abridged and updated version of the Basic Protocols from our original protocol paper, which can be downloaded here: OPTiKD
Some plasmids can be obtained from Addgene (https://www.addgene.org/Ludovic_Vallier/); contact us for the Zinc Finger Nucleases plasmids
Some plasmids can be obtained from Addgene (https://www.addgene.org/Ludovic_Vallier/); contact us for the Zinc Finger Nucleases plasmids
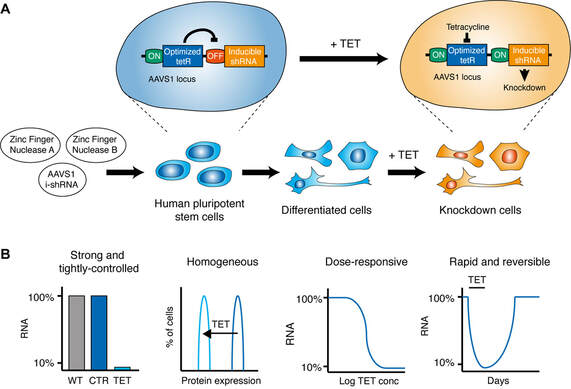
Figure 1. The optimized inducible knockdown (OPTiKD) system. (A) Schematic of the generation of OPTiKD human pluripotent stem cells (hPSCs) and of the resulting genetic circuit which allows tetracycline (TET)–dependent induction of gene knockdown both in hPSCs and hPSC-derived cells. (B) Schematic of the properties of OPTiKD.
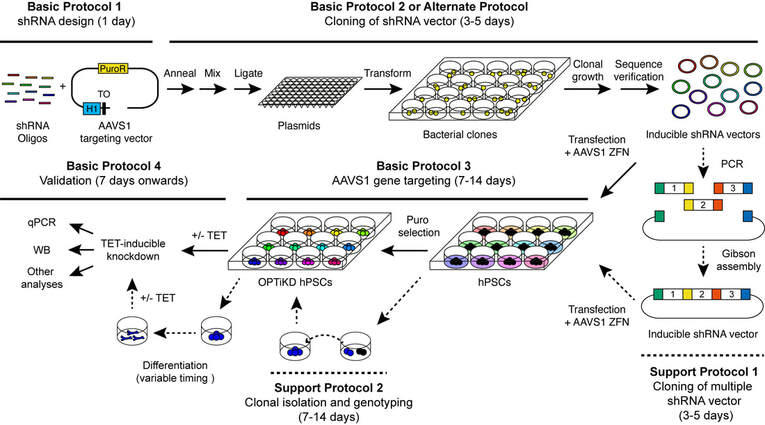
Figure 2. Generation and characterization of OPTiKD hPSCs. Schematic overview of the procedure detailing how the various protocols relate to each other. Optional steps are indicated by dashed arrows. PuroR: puromycin resistance; H1: H1 Pol III promoter; TO: tet operator; ZFN: zinc-finger nucleases. Refer to the text for other common abbreviations.
Basic Protocol 1: Design of Oligonucleotides for shRNA Cloning
The first step in generating OPTiKD hPSCs consists in obtaining oligonucleotides to be used to clone the shRNA sequence in the targeting vector. First, a suitable shRNA sequence must be identified. Second, oligonucleotides are designed to add appropriate 5’- and 3’-end overhangs to facilitate directional cloning.
1.A: shRNA design
Identifying a good shRNA is the most critical parameter when generating OPTiKD hPSCs. Validated shRNA sequences can be obtained from various sources such as previous reports in the scientific literature, public shRNA databases, or commercial suppliers. Alternatively, shRNAs can be designed in house by using available algorithms (reviewed in Fakhr, Zare, & Teimoori-Toolabi, 2016). For genes that have no reported validated shRNA, we advise investigators to take advantage of the RNAi Consortium shRNA library (also known as the TRC library or the Genetic Perturbation Platform: https://portals.broadinstitute.org/gpp/public/; Moffat et al., 2006), which contains pre-designed shRNAs for all human genes, >50% of which have been experimentally validated. Such validation data is available from SigmaAldrich, one of the commercial distributors of the TRC library (https://www.sigmaaldrich.com/life-science/functional-genomics-and-rnai/shrna/individual-genes.html).
This basic protocol assumes that an shRNA from the TRC library is being used. When using alternative sources for an shRNA, ignore steps 1, 2, and 3 of this protocol and proceed directly to step 4. The OPTiKD method has been successfully used with a variety of shRNA designs (Bertero et al., 2016). When designing the shRNA in house, make sure to follow appropriate design rules according to the most recent literature on the topic (reviewed in Fakhr et al., 2016). Further, it is crucial to avoid shRNAs containing a poly(T) tract longer than four nucleotides, as this could lead to premature shRNA termination (TRC library shRNA are pre-screened to avoid this problem). We generated an electronic worksheet file that can be used to perform all the oligonucleotide design steps (steps 4 to 6) in an automatic fashion; we recommend using such tool once familiar with the design principles described here. The worksheet can be downloaded from https://docs.google.com/spreadsheets/d/18jzVw-5kHzw8xAXkvvFYZizL-y5Hapb1r4Fp6zTXk4U/edit?usp=sharing.
1.B: Design of oligonucleotides for cloning the shRNA into the targeting vector
The cloning procedure will be described in more detail in Basic Protocol 2. Briefly, two single-stranded oligonucleotides are annealed to form a double-stranded DNA sequence containing the shRNA sequence followed by a Pol III terminator (consisting of a stretch of four or more thymidines), as well as 5’- and 3’-end “sticky” overhangs that are complementary to those generated in the targeting vector following restriction digestion with BglII and SalI (Figures 3E and 4C). This design facilitates directional cloning of the shRNA into the targeting vector. Furthermore, the oligonucleotides are designed to destroy the BglII restriction enzyme site in the final plasmid in order to allow screening of correct recombinant clones by diagnostic restriction digestion (Figure 4D). As transcription from the H1 Pol III promoter is more efficient and precise when a guanine (“G”) or an adenosine (“A”) base is found on the +1 site (Ma et al., 2014; Ranganathan, Wahlin, Maruotti, & Zack, 2014), the shRNA is modified to satisfy this criteria if needed.
The cloning procedure will be described in more detail in Basic Protocol 2. Briefly, two single-stranded oligonucleotides are annealed to form a double-stranded DNA sequence containing the shRNA sequence followed by a Pol III terminator (consisting of a stretch of four or more thymidines), as well as 5’- and 3’-end “sticky” overhangs that are complementary to those generated in the targeting vector following restriction digestion with BglII and SalI (Figures 3E and 4C). This design facilitates directional cloning of the shRNA into the targeting vector. Furthermore, the oligonucleotides are designed to destroy the BglII restriction enzyme site in the final plasmid in order to allow screening of correct recombinant clones by diagnostic restriction digestion (Figure 4D). As transcription from the H1 Pol III promoter is more efficient and precise when a guanine (“G”) or an adenosine (“A”) base is found on the +1 site (Ma et al., 2014; Ranganathan, Wahlin, Maruotti, & Zack, 2014), the shRNA is modified to satisfy this criteria if needed.
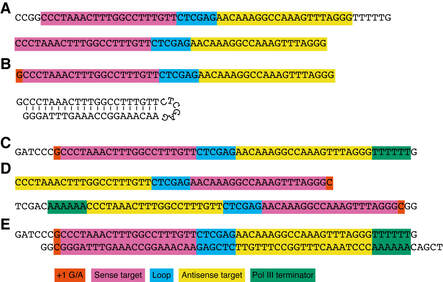
Figure 3. Design of shRNAs for OPTiKD. Example of the design procedure described in Basic Protocol 1. The relevant sequence elements are color-coded. +1G/A:guanine/adenosine found on the first translated nucleotide following cloning into the H1 promoter. (A) Sequence from the TRC shRNA database as obtained from the Sigma-Aldrich Web site (top) and after trimming of extra nucleotides (shRNA proper, bottom). (B) shRNA modified to start with a guanine to ensure maximal activity of the H1 promoter. The predicted hairpin secondary structure is shownonthe bottom. (C) Top oligonucleotide for shRNA cloning. (D) Reverse complement of the shRNA in panel B (top), and bottom oligonucleotide for shRNA cloning (bottom). (E) Predicted double-stranded DNAobtained after annealing of the top and bottom oligonucleotides from panels C and D.
Materials:
- SnapGene software (GSL Biotech LLC)
Procedure:
1. Obtain a shRNA target sequence for the gene of interest from the TRC library (https://portals.broadinstitute.org/gpp/public/gene/search).
Search the gene of interest by its official gene symbol: a list of all available shRNAs will appear and can be visualized online or downloaded as a comma-separated (CSV) file. In this list, identify the 20- to 21-bp-long target sequence (sense shRNA target site).
2. Check that the 21-bp sense shRNA target site maps to the desired transcript by using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
A 100% match is to be expected. The shRNAs from the TRC library have been designed to target all annotated isoforms of a given gene, but as annotations are ever evolving it is possible that the isoform of interest might not be targeted by the selected shRNA. If so, go back to step 1 and select a new shRNA.
3. Design the shRNA sequence by joining the following segments in 5’ to 3’ orientation
- The sense shRNA target site identified in step 1.
- The 6-bp “loop” sequence 5’-CTCGAG-3’.
- The reverse complement of the sense shRNA target site identified in step 1 (antisense shRNA target site).
Note that the shRNAs listed on the Sigma-Aldrich Web site will most likely have initial “CCGG” and final “TTTTTG/TTTTG” sequences, which must be ignored as these are not part of the shRNA proper. When the shRNA sequence is transcribed by Pol III, it will form a hairpin structure with the complementary sense and antisense target sites forming a stem through Watson-Crick base pair interactions, and an extruding 6-bp loop (Figure 3B).
4. Check to see if the sense shRNA target site starts with a guanine (“G”) or an adenosine (“A”): if not, insert a “G” at the beginning of the sense shRNA target site (Figure 3B).
Note that a complementary final “C” should not be added to the antisense shRNA target site, as the extra “G” hanging on the 5-end of the shRNA will not create any issue and it is better to preserve the structure of the 20- to 21-bp-long hairpin.
5. Design the “top” oligonucleotide by:
- Taking the shRNA sequence from step 4.
- Adding 5’-GATCCC-3’ to the 5’-end.
- Adding 5’-TTTTTTG-3’ to the 3’-end (Figure 3C).
6. Design the “bottom” oligonucleotide by:
- Obtaining the reverse complement of the shRNA sequence from step 4.
- Adding 5’-TCGACAAAAAA-3’ to the 5’-end.
- Adding 5’-GG-3’ to the 3’ end (Figure 3D).
7. Optional: Use SnapGene software for in silico simulation of the subsequent cloning step by:
- Annealing the oligonucleotides.
- Digesting pAAV-Puro_siKD with BglII and SalI (for the sequence see Materials in Basic Protocol 2).
- Ligating the two fragments.
We strongly recommend this step to verify that the oligonucleotides are predicted to anneal perfectly and form a structure with theoverhangs required for the subsequent cloning step (Figure 3E). Furthermore, the resulting plasmid map will be used to facilitate the ensuing analysis of sequencing results of plasmid clones. If SnapGene is unavailable, use equivalent software for in silico analysis of DNA sequences and simulation of molecular cloning procedures.
8. Order top and bottom oligonucleotides as desalted purified products from SigmaAldrich or from an alternative preferred commercial supplier.
A production scale of 0.05 µmol is sufficient. PAGE purification of the oligonucleotides is not required in our experience but could be beneficial if the subsequent cloning step proves inefficient.
Basic Protocol 2: Generation of Inducible shRNA Targeting Vector
In this second part of the procedure, the oligonucleotides obtained from Basic Protocol 1 are used to generate an AAVS1 targeting vector for inducible shRNA expression. First, the oligonucleotides are annealed, phosphorylated, and ligated to a pre-digested plasmid. Secondly, the recombinant DNA is transformed into E. coli for clonal propagation. Finally, bacterial clones are screened to identify correct recombinants that will be used for plasmid preparation and subsequent gene targeting.
Basic Protocol 2: Generation of Inducible shRNA Targeting Vector
In this second part of the procedure, the oligonucleotides obtained from Basic Protocol 1 are used to generate an AAVS1 targeting vector for inducible shRNA expression. First, the oligonucleotides are annealed, phosphorylated, and ligated to a pre-digested plasmid. Secondly, the recombinant DNA is transformed into E. coli for clonal propagation. Finally, bacterial clones are screened to identify correct recombinants that will be used for plasmid preparation and subsequent gene targeting.
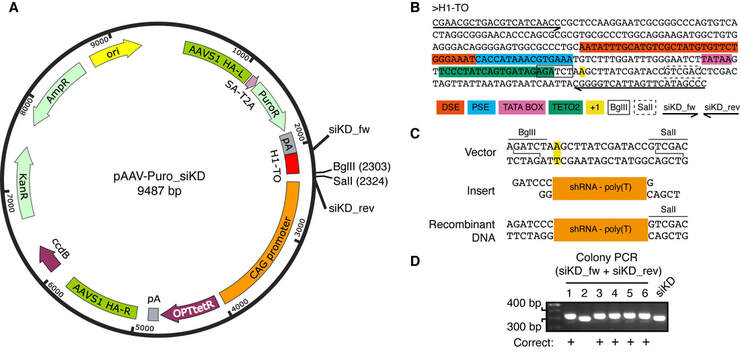
Figure 4. Cloning of shRNAs into the OPTiKD plasmid. (A) Map of the plasmid used to generate OPTiKD hPSCs. The position of restriction sites and primers relevant to the protocol is shown. HA-L/R: left/right homology arm; SA: splice acceptor; T2A: self-splicing T2A sequence; PuroR: puromycin resistance cDNA; pA: polyadenylation sequence; H1-TO: tetracycline-inducible H1 RNA Polymerase III promoter containing a tet operator; CAG: CMV early enhancer, chicken β-actin and rabbit β-globin hybrid promoter; OPTtetR: codon-optimized tetracycline-responsive repressor protein cDNA; ccdB: ccdB toxin (truncated and thus inactive); KanR: kanamycin resistance; AmpR: ampicillin resistance; ori: high-copy origin of replication. (B) Nucleotide sequence of the tetracycline-inducible H1-TO RNA Polymerase III promoter. Key sequence features are color coded, and the locations of primers are shown with arrows indicating their 5'-3' directionality. The restriction enzyme cutsites used for shRNA cloning are shownin boxes. DSE: distal sequence element; PSE: proximal sequence element; TETO2: tet operator; +1: start position of RNA transcription. (C) Schematics of the shRNA cloning procedure described in Basic Protocol 2. The portion of the vector being cutisshown on top, and the restriction enzyme sites and resulting overhangs are indicated. The +1 position is in yellow (panel B). The double-stranded oligonucleotide containing the shRNA (Figure 3) is schematized in the middle. The product resulting from ligation of the cut vector and insert is on the bottom. Note that the BglII site is destroyed after ligation. (D) Representative example of colony PCR results from six screened bacterial clones. Clones with the expected larger amplification product are indicated. siKD: control PCR performed on pAAV-Puro_siKD (indicating the size of the empty vector).
2.A: The OPTiKD plasmid
This OPTiKD method is based on usage of pAAV-Puro_siKD (Figure 4A), a bacterial plasmid which allows selective gene targeting of an all-in-one tetracycline inducible shRNA cassette into the human AAVS1 locus (PPP1R12C gene, located on chromosome 19). The gene targeting step is described in detail in Basic Protocol 3. Briefly, this is achieved by the generation of a genomic DNA double-strand break by obligate heterodimer zinc-finger nucleases (ZFN) that target the first intron of PPP1R12C (Bertero et al., 2016; Hockemeyer et al., 2009). Such DNA damage is resolved via homologous directed-repair (HDR) based on the presence of two homology arms on pAAV-Puro_siKD that map to the 5’- and 3’-end of the double-strand break. This leads to the insertion of the DNA cassette present between the 5’- and 3’-end homology arms. This DNA cassette contains an antibiotic drug resistance gene trap that facilitates the selection of gene targeted hPSCs, and is followed by the all-in-on-inducible shRNA cassette proper.
The antibiotic drug resistance gene used is puromycin-N-acetyltransferase (PAT), which inactivates the protein synthesis inhibitor puromycin, a drug commonly used to rapidly and specifically select eukaryotic cells. The PAT gene is driven by the endogenous PPP1R12C promoter through a gene-trap approach rather than relying on an autonomous promoter (Hockemeyer et al., 2009). This strategy relies on a strong splice acceptor that “hijacks” the PPP1R12C transcripts and splices in the PAT cDNA. This design strongly increases the probability that drug-resistant hPSCs are the result of correct gene targeting to the AAVS1 locus. Indeed, random integration of the pAAV-Puro_siKD plasmid will most often not result in expression of the PAT gene due to the lack of a dedicated promoter (the exception to this is if the plasmid is integrated in an intronic region of an expressed gene, but this is a rare event that in our experience occurs with less than 5% frequency; see Basic Protocols 3 and 4). Note that random integration of the targeting plasmid is an event that happens with comparable if not higher frequency to site-specific gene targeting through HDR. Therefore, the use of a gene-trap approach for the drug resistance gene is key to obtaining correctly targeted cells with high frequency (in our experience this is more than 95%; see Basic Protocols 3 and 4).
The inducible shRNA is placed under the transcriptional control of an H1 PolIII promoter modified to include a tet operator sequence (TO) after the TATA box (H1-TO promoter, Figure 4B; Brummelkamp, Bernards, & Agami, 2002). Under control conditions (absence of the drug tetracycline) this promoter is efficiently bound by the tetR protein, which prevents expression of the shRNA. The tetR protein is expressed from the same targeting vector and is under the transcriptional control of the strong constitutive CAG promoter, which is highly expressed and resistant to silencing in hPSCs and differentiated cell types. The tetR protein was subjected to multi-parameter codon and RNA optimization for expression in human cells (Fath et al., 2011), which increased its steady-state expression by approximately one order of magnitude (Bertero et al., 2016). We named the resulting product OPTtetR, for codon OPTimized tetR, and demonstrated that when expressed in the all-in-one inducible shRNA OPTiKD cassette it was sufficient to virtually abolish “leaky” expression of the shRNA in the absence of tetracycline, a problem which was shown to markedly plague traditional TET-OFF-inducible shRNA methods based on the wild-type bacterial tetR (Bertero et al., 2016; Henriksen et al., 2007). We also showed that the OPTtetR is fully derepressible by addition of tetracycline, thus maximizing the dynamic range of inducible knockdown. The beneficial effects of using the OPTtetR are most likely due to its higher protein expression, which is expected to ensure higher binding efficiency to the cognate TO site and thus more consistent repression (Gray et al., 2007). Note that in contrast to the PAT gene the OPTtetR expression is driven by a constitutive promoter independent from the integration site. This strategy ensures stable expression even upon differentiation of hPSCs into lineages that might show low transcriptional activity of the PPP1R12C gene. A final point of note is that the orientation of the inducible shRNA and constitutive OPTtetR expression cassettes is key to the functionality of the all-in-one approach described here (A. Bertero unpub. observ.).
Transcription of the two PolII-dependent transgenes (PAT and OPTtetR) is terminated by the strong bovine growth hormone polyadenylation signal, while the shRNA is terminated by a poly(T) tract (see Section 1.B in the introduction to Basic Protocol 1). The plasmid backbone contains a pUC bacterial origin of replication for high-copy plasmid production in E. coli, and an ampicillin resistance gene to allow selective propagation of bacteria containing the recombinant plasmid (a kanamycin resistance gene is also present but not used for the procedure described here). Note that a partial ccdB gene (which encodes for a bacterial toxin that poisons DNA gyrase) is present on the backbone, but this is not functional, as it lacks a start codon (this represents a remnant from the original backbone used to generate pAAV-Puro_siKD). Therefore, usage of special E. coli strains that tolerate expression of the ccdB toxin is not required for propagation of the plasmid.
2.B: Cloning of shRNA into the OPTiKD plasmid
The shRNA is cloned into pAAV-Puro_siKD by means of a simplified restriction enzyme–based molecular cloning step. The plasmid is cut with two different restriction enzymes (BglII and SalI), thus generating distinct and non-compatible sticky 5’- and 3’-end overhangs that allow directional cloning (Figure 4C). As single- and double-cut plasmids cannot be distinguished based on their size by traditional agarose gel DNA electrophoresis, the digested plasmid is also dephosphorylated. This is done to reduce the rate of self-ligation of contaminating molecules in which only one of the two restriction sites was successfully digested (and that are thus presenting compatible sticky ends). This means that the double-stranded DNA encoding for the shRNA obtained from annealed oligonucleotides must be phosphorylated prior to ligation. As mentioned in Section 1.B (see introduction to Basic Protocol 1), the BglII site is destroyed upon successful ligation, which facilitates screening of recombinant plasmids by diagnostic restriction digestion (Figure 4C). Following successful ligation, the shRNA sequence will be placed so that the first base to be transcribed (a guanine or alanine; see Section 1.B in the introduction to Basic Protocol 1) is on the +1 position (indicating the known transcriptional start site of the H1 promoter, which is 26 bp downstream the end of the TATA box; Figure 4B). The poly(T) stretch after the shRNA sequence functions as transcriptional terminator for Pol III. Recombinant plasmids isolated from bacterial clones are screened by colony PCR and/or diagnostic restriction digestion, and correct ligation products are further screened by Sanger sequencing to confirm that no mutations are present in the shRNA sequence. The inducible shRNA plasmid is finally expanded into E. coli in preparation to the gene targeting step (Basic Protocol 3).
The shRNA is cloned into pAAV-Puro_siKD by means of a simplified restriction enzyme–based molecular cloning step. The plasmid is cut with two different restriction enzymes (BglII and SalI), thus generating distinct and non-compatible sticky 5’- and 3’-end overhangs that allow directional cloning (Figure 4C). As single- and double-cut plasmids cannot be distinguished based on their size by traditional agarose gel DNA electrophoresis, the digested plasmid is also dephosphorylated. This is done to reduce the rate of self-ligation of contaminating molecules in which only one of the two restriction sites was successfully digested (and that are thus presenting compatible sticky ends). This means that the double-stranded DNA encoding for the shRNA obtained from annealed oligonucleotides must be phosphorylated prior to ligation. As mentioned in Section 1.B (see introduction to Basic Protocol 1), the BglII site is destroyed upon successful ligation, which facilitates screening of recombinant plasmids by diagnostic restriction digestion (Figure 4C). Following successful ligation, the shRNA sequence will be placed so that the first base to be transcribed (a guanine or alanine; see Section 1.B in the introduction to Basic Protocol 1) is on the +1 position (indicating the known transcriptional start site of the H1 promoter, which is 26 bp downstream the end of the TATA box; Figure 4B). The poly(T) stretch after the shRNA sequence functions as transcriptional terminator for Pol III. Recombinant plasmids isolated from bacterial clones are screened by colony PCR and/or diagnostic restriction digestion, and correct ligation products are further screened by Sanger sequencing to confirm that no mutations are present in the shRNA sequence. The inducible shRNA plasmid is finally expanded into E. coli in preparation to the gene targeting step (Basic Protocol 3).
Materials:
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2001)
2.C: Preparation of cloning vector
1. One-step digest and dephosphorylate pAAV-Puro_siKD by preparing the following reaction:
A shorter incubation of 5 to 30 min can alternatively be performed, as these enzymes are fast cutters, but we recommend 1 hr tomaximize efficiency since a large amount of plasmid is being digested. No significant restriction enzyme star activity should beobserved for up to 3 hr of incubation, and a longer incubation time is not recommended. The above reaction can be scaled down to digest 1 to 2 µg of plasmid if desired. However, we find it is best to prepare a larger batch of plasmid to be used for multiple ligations.
2. Add 18 µl of gel loading dye, mix and run on a 1% (w/v) agarose gel in TBE containing 0.5 μg/ml ethidium bromide. Include the DNA ladder in one separate well as molecular weight control. Visualize the DNA using a UV transilluminator, excise the 9466-bp band with a clean scalpel, and perform DNA gel extraction using QIAEXII Gel Extraction Kit according to the manufacturer’s instructions and aiming for a final concentration of 25 to 100 ng/µl.
Essential protocols for agarose gel electrophoresis are included in Voytas (2001). It is advisable to run 500 ng of uncut plasmid in a separate well as control for complete digestion. Should undigested plasmid be contaminating the preparation, we advise repeating step 1 with a longer incubation time and/or reducing the amount of plasmid being digested. When a large plasmid batch is being prepared, divide the gel-extracted vector in 5- to 10-µl aliquots and store for later use at –20°C for at least 6 months. Ethidium bromide is a known carcinogen and should be handled with care while wearing appropriate personal protective equipment and in accordance with local safety regulations. Alternative products carrying lower risk for carcinogenesis are available on the market and are an advisable alternative wherever possible.
2.D: Preparation of shRNA inserts
3. Assemble the following reaction in a PCR tube:
4. Phosphorylate, denature, and anneal the shRNA oligonucleotides by incubating the reaction from step 3 in a thermocycler according to the following program (lid kept at 95°C):
5. Optional: Verify the annealing by running 10 pmol of the annealed double-stranded oligonucleotides side-by-side to single-stranded oligonucleotides (mixed as described in step 3 ) on a high-percentage (4%, w/v) agarose gel (use low-melting-point agarose; also see step 2).
The annealed oligonucleotides should show a band at a higher molecular weight compared to the single-stranded oligonucleotides. Note that single-strand oligonucleotides will not resolve at the expected size, as they form secondary structures under non denaturing conditions. Estimate the proportion of annealed versus non-annealed oligonucleotides: this should be >70%. Repeat steps 3 to 4 if necessary.
6. Dilute the annealed double-stranded oligonucleotides from step 3 1: 500 in ddH2O by performing serial dilutions of 1:10 and 1:50. Store on ice.
The diluted double-stranded oligonucleotides are not stable and should be stored on ice or at 4°C for not more than 1 hr.
2.E: Generation of recombinant DNA
7. Assemble the following ligation reaction in a PCR tube using the Rapid DNA Ligation Kit:
Longer incubation times are usually not required, but overnight incubation at room temperature can be attempted if the ligation according to this protocol proves inefficient. An additional negative-control ligation omitting the oligo (all the reagents above but ’b’, its volume to be replaced by ddH2O) can help in determining the success of the molecular cloning experiment, and is recommended for first-time user or as a troubleshooting experiment.
8. Transform 5 µl of the ligation product from step 7 into 50 µl ofα-select E. coli according to manufacturer’s instructions using a heated water bath for heat shock.
If preferred, use an equivalent highly competent and recombinase-deficient strain suitable for plasmid propagation. Recovery of transformed bacteria at 37°C in SOC medium is optional when using plasmids carrying an ampicillin-resistance gene, but we strongly advise investigators to perform it, as it has been shown to improve the yield by at least a factor of 2. Use a minimal volume of medium (100 to 250 µl) to facilitate bacterial plating in step 9.
9. Plate all of the transformed bacteria onto a previously prepared LB agar plate containing 100 µg/ml ampicillin and incubate overnight at 37°C in a humidified incubator.
To facilitate the spreading of all transformed bacteria, we advise briefly centrifuging the tube for 15 sec at 6000 × g, room temperature, discarding the supernatant by gently pouring, and resuspending the bacterial pellet in the remaining medium (50 to 100 µl) before plating.
- pAAV-Puro_siKD plasmid (Addgene, cat. no. 86695)
- FastDigest BglII (ThermoFisher, cat. no. FD0083)
- FastDigest SalI (ThermoFisher, cat. no. FD0644)
- FastAP Thermosensitive Alkaline Phosphatase (1 U/µl; ThermoFisher, cat. no. EF0654)
- 10x FastDigest Green Buffer (ThermoFisher, cat. no. B72)
- Ultrapure DNase/RNase-free distilled water (ThermoFisher, cat. no. 10977015)
- 6x gel loading dye (New England Biolabs, cat. no. B7024S)
- 1% (w/v) agarose gel in TBE containing 0.5 μg/ml ethidium bromide (see recipe; use molecular biology grade agarose; ThermoFisher, cat. no. 17850)
- 1 Kb plus DNA ladder (ThermoFisher, cat. no. 10787018)
- QIAEX II Gel Extraction Kit (Qiagen, cat. no. 20021)
- “Top” single-stranded oligonucleotide for shRNA (custom oligonucleotide; Basic Protocol 1, step 8)
- “Bottom” single-stranded oligonucleotide for shRNA (custom oligonucleotide; Basic Protocol 1, step 8)
- T4 DNA Ligase Reaction Buffer (New England Biolabs, cat. no. B0202S)
- T4 Polynucleotide Kinase (New England Biolabs, cat. no. M0201S)
- 4% (w/v) agarose gel in TBE containing 0.5 μg/ml ethidium bromide (see recipe; use UltraPure Low Melting Point Agarose; ThermoFisher, cat. no. 16520050)
- Rapid DNA Ligation Kit (ThermoFisher, cat. no. K1422)
- α-select Gold Efficiency E. coli (Bioline, cat. no. BIO-85027)
- SOC medium (ThermoFisher, cat. no. 15544034)
- LB agar plates containing 100 µg/ml ampicillin (see recipe)
- siKD_fw primer (custom oligonucleotide: 5'-CGAACGCTGACGTCATCAACC-3')
- siKD_rev primer (custom oligonucleotide: 5'-GGGCTATGAACTAATGACCCCG-3')
- dNTP mix (Promega, cat. no. U1511)
- GoTaq Flexi DNA Polymerase (Promega, cat. no. M8291)
- Luria Bertani (LB) broth containing 100 µg/ml ampicillin (see recipe)
- 1.5% (w/v) agarose gel in TBE containing 0.5 μg/ml ethidium bromide (see recipe; use standard molecular biology grade agarose)
- QIAprep Spin Miniprep Kit (Qiagen, cat. no. 27104)
- Glycerol for molecular biology, >99% (Sigma-Aldrich, cat. no. G5516-100ML)
- QIAfilter Plasmid Midi Kit (Qiagen, cat. no. 12243)
- UV transilluminator
- PCR tubes
- Thermocycler with heated lid
- Heated water bath
- Humidified bacterial incubator
- Sanger sequencing facility (or commercial provider)
- Bacterial culture orbital shaker
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2001)
2.C: Preparation of cloning vector
1. One-step digest and dephosphorylate pAAV-Puro_siKD by preparing the following reaction:
- pAAV-Puro_siKD, variable volume (5 µg)
- FastDigest BglII, 3 µl
- FastDigest SalI, 3 µl
- FastAP (1 U/µl), 3 µl
- FastDigest Green Buffer (10x), 9 µl
- Ultrapure ddH2O to 90 µl
A shorter incubation of 5 to 30 min can alternatively be performed, as these enzymes are fast cutters, but we recommend 1 hr tomaximize efficiency since a large amount of plasmid is being digested. No significant restriction enzyme star activity should beobserved for up to 3 hr of incubation, and a longer incubation time is not recommended. The above reaction can be scaled down to digest 1 to 2 µg of plasmid if desired. However, we find it is best to prepare a larger batch of plasmid to be used for multiple ligations.
2. Add 18 µl of gel loading dye, mix and run on a 1% (w/v) agarose gel in TBE containing 0.5 μg/ml ethidium bromide. Include the DNA ladder in one separate well as molecular weight control. Visualize the DNA using a UV transilluminator, excise the 9466-bp band with a clean scalpel, and perform DNA gel extraction using QIAEXII Gel Extraction Kit according to the manufacturer’s instructions and aiming for a final concentration of 25 to 100 ng/µl.
Essential protocols for agarose gel electrophoresis are included in Voytas (2001). It is advisable to run 500 ng of uncut plasmid in a separate well as control for complete digestion. Should undigested plasmid be contaminating the preparation, we advise repeating step 1 with a longer incubation time and/or reducing the amount of plasmid being digested. When a large plasmid batch is being prepared, divide the gel-extracted vector in 5- to 10-µl aliquots and store for later use at –20°C for at least 6 months. Ethidium bromide is a known carcinogen and should be handled with care while wearing appropriate personal protective equipment and in accordance with local safety regulations. Alternative products carrying lower risk for carcinogenesis are available on the market and are an advisable alternative wherever possible.
2.D: Preparation of shRNA inserts
3. Assemble the following reaction in a PCR tube:
- Top oligo (200 µM in ddH2O) (Basic Protocol 1), 5 µl
- Bottom oligo (200 µM in ddH2O) (Basic Protocol 1), 5 µl
- T4 DNA Ligase Reaction Buffer (10×), 2 µl
- T4 Polynucleotide Kinase, 1 µl
- Ultrapure ddH2O, 7 µl
4. Phosphorylate, denature, and anneal the shRNA oligonucleotides by incubating the reaction from step 3 in a thermocycler according to the following program (lid kept at 95°C):
- 37°C, 1 hr
- 95°C, 5 min
- Ramp down to 80°C at –0.1°C/sec:
- 80°C, 4 min
- Ramp down to 75°C at –0.1°C/sec:
- 75°C, 4 min
- Ramp down to 70°C at –0.1°C/sec:
- 70°C, 4 min
- Ramp down to 10°C at –0.1°C/sec:
- 10°C, hold
5. Optional: Verify the annealing by running 10 pmol of the annealed double-stranded oligonucleotides side-by-side to single-stranded oligonucleotides (mixed as described in step 3 ) on a high-percentage (4%, w/v) agarose gel (use low-melting-point agarose; also see step 2).
The annealed oligonucleotides should show a band at a higher molecular weight compared to the single-stranded oligonucleotides. Note that single-strand oligonucleotides will not resolve at the expected size, as they form secondary structures under non denaturing conditions. Estimate the proportion of annealed versus non-annealed oligonucleotides: this should be >70%. Repeat steps 3 to 4 if necessary.
6. Dilute the annealed double-stranded oligonucleotides from step 3 1: 500 in ddH2O by performing serial dilutions of 1:10 and 1:50. Store on ice.
The diluted double-stranded oligonucleotides are not stable and should be stored on ice or at 4°C for not more than 1 hr.
2.E: Generation of recombinant DNA
7. Assemble the following ligation reaction in a PCR tube using the Rapid DNA Ligation Kit:
- Cut vector from step 2, variable volume (50 ng)
- Diluted oligo from step 6, 4 µl
- 5× Rapid Ligation Buffer, 2 µl
- T4 DNA Ligase, 5 U/µl, 1 µl
- Ultrapure ddH2O to 10 µl
Longer incubation times are usually not required, but overnight incubation at room temperature can be attempted if the ligation according to this protocol proves inefficient. An additional negative-control ligation omitting the oligo (all the reagents above but ’b’, its volume to be replaced by ddH2O) can help in determining the success of the molecular cloning experiment, and is recommended for first-time user or as a troubleshooting experiment.
8. Transform 5 µl of the ligation product from step 7 into 50 µl ofα-select E. coli according to manufacturer’s instructions using a heated water bath for heat shock.
If preferred, use an equivalent highly competent and recombinase-deficient strain suitable for plasmid propagation. Recovery of transformed bacteria at 37°C in SOC medium is optional when using plasmids carrying an ampicillin-resistance gene, but we strongly advise investigators to perform it, as it has been shown to improve the yield by at least a factor of 2. Use a minimal volume of medium (100 to 250 µl) to facilitate bacterial plating in step 9.
9. Plate all of the transformed bacteria onto a previously prepared LB agar plate containing 100 µg/ml ampicillin and incubate overnight at 37°C in a humidified incubator.
To facilitate the spreading of all transformed bacteria, we advise briefly centrifuging the tube for 15 sec at 6000 × g, room temperature, discarding the supernatant by gently pouring, and resuspending the bacterial pellet in the remaining medium (50 to 100 µl) before plating.
2.F: Colony PCR screening of bacterial clones
10. Prepare the following PCR master mix using GoTaq Flexi DNA Polymerase (volumes for one reaction; adjust as needed and include one extra sample for a negative control):
Screening of six to eight colonies per construct is recommended. If a negative control ligation was performed during step 7, this can be used to estimate the level of background due to self-ligated vector. If this is really low, screening of two to four colonies should be sufficient. Since the efficiency of shRNA cloning for most sequences is very high (>90%), colony PCR screening might be entirely bypassed (skip steps 10 to 15). If this is the case, it is recommended to isolate six to eight clones for plasmid miniprep and confirm successful cloning by BglII digest followed by DNA sequencing (steps 16 to 18).
11. For each clone to be analyzed, prepare two sets of sterile PCR tubes containing 200 µl of LB broth with 100 µg/ml of ampicillin,and 10 µl of ultrapure ddH2O, respectively (use a 96 well plate if analyzing a large number of clones).
12. Using a micro-pipette set on 5 µl and a sterile tip with an aerosol-barrier filter, pick a single bacterial colony from the plate from step 9. Pipet up and down five times into the tube with ddH2O to break down the colony, transfer 5 µl of the bacterial suspension to the PCR tube, and pipet the remaining 5 µl to the tube with LB broth. Change tip and repeat this step for all clones to be analyzed. At the end of this step transfer the tubes with LB broth to 4°C storage for later use.
13. To the tube prepared for the negative control, add 5 µl of uncut pAAV-Puro_siKD plasmid diluted to 0.2ng/µl. This reaction will facilitate determining positive clones based on the size of the PCR band (step 15).
14. Perform the PCR according to the following protocol:
15. Run 7 µl of the PCR reaction on a 1.5% (w/v) agarose gel containing 0.5 μg/ml ethidium bromide. Run at 100 V for at least 30 min, image the gel using an UV transilluminator, and identify the clones giving a product which size is approximately 350 bp (depending on the size of the shRNA sequence; Figure 4D).
Self-ligated vectors will result into a product of 295 bp, which can be identified from the negative control from step 13 (Figure 4D). If the size difference between the shRNA containing plasmids and the empty control vector is unclear, run the gel for a longer time until the size resolution is sufficient.
16. Inoculate the bacterial clones containing the correct ligation product using the liquid culture from step 12 by transferring 100 µl of the bacterial suspension to a microbiology tube containing 4 ml of LB broth with 100 µg/ml of ampicillin. Incubate in a bacterial culture orbital shaker at 37°C overnight (16 hr) while shaking at 225 rpm.
We recommend expanding two to four clones in order to have some backup should the subsequent sequencing results demonstrate any mutation in the shRNA sequence (step 19). Store the bacterial plate from step 9 at 4°C for up to 2 weeks. This can be reused to repeat steps 12 to 15 if required.
2G: Isolation and quality control of the plasmid
17. Isolate plasmids from bacterial cultures (step 16) using the QIAprep Spin Miniprep Kit following manufacturer’s instructions. Save 200 µl of the bacterial culture at 4°C.
18. Optional: Confirm shRNA cloning by performing a diagnostic restriction digestion with BglII using pAAV-Puro_siKD as control.
Correct recombinants containing the shRNA will not be linearized by BglII (provided no new BglII site is found in the shRNA sequence), as this restriction site is destroyed during the cloning. If the plasmids have been pre-screened by colony PCR (steps 10 to 15), this step is redundant and can be omitted.
19. Perform Sanger DNA sequencing using the siKD_fw primer to confirm the presence of the shRNA and its correct sequence.
The strong hairpin secondary structure of certain shRNAs can lead to difficulties in Sanger sequencing. In this case, try sequencing with siKD_rev primer and ask the sequencing company to use conditions appropriate for sequencing of GC-rich templates or sequences with a strong secondary structure. We recommend using the plasmid map generated in step 7 of Basic Protocol 1 as reference to align the sequencing results.
20. Using the liquid culture from step 17, expand the bacterial clones containing the correct shRNA in 50 ml of LB broth with 100 µg/ml of ampicillin. Incubate in a bacterial culture orbital shaker at 37°C overnight (16 hr) while shaking at 225 rpm.
The liquid culture from step 17 should be used within a week of storing at 4°C. Otherwise we recommend re-transforming the plasmid from step 17, picking a fresh bacterial colony, pre-inoculating it in 2 ml of LB broth with 100 µg/ml of ampicillin for 6 hr, and then diluting it 1:1000 in 50 ml of LB broth to be incubated as described above.
21. Prepare a glycerol stock to be stored at –80°C for long-term back up by mixing 200µl of sterile autoclaved 50%(v/v) glycerol in ddH2O with 200 µl of the bacterial culture from step 20.
Glycerol stocks can be stored for up to 10 years and used to recover the plasmid. For this, streak the glycerol stock as in step 9 then repeat step 20.
22. Use the remaining culture to isolate the plasmid using the QIAfilter Midiprep Kit. Resuspend the purified plasmid in elution buffer (10 mM Tris·Cl, pH 7.5 in ddH2O) at a concentration of 500 to 1000 ng/µl and store at –20°C for up to a year.
Avoid repeated freeze-thaw cycles of the plasmid. ddH2O can be used instead of elution buffer, but the shelf life of the plasmids will decrease. We did not notice interference of elution buffer with the subsequent gene targeting steps. We advise using freshly prepared plasmids (less than a month of storage) to increase the efficiency of gene targeting. We strongly advise repeating step 19 to confirm the shRNA sequence before proceeding with gene-targeting experiments.
10. Prepare the following PCR master mix using GoTaq Flexi DNA Polymerase (volumes for one reaction; adjust as needed and include one extra sample for a negative control):
- siKD_fw primer (10 µM in ddH2O), 0.5 µl
- siKD_rev primer (10 µM in ddH2O), 0.5 µl
- dNTP mix (10 mM), 0.5 µl
- MgCl2 (25 mM), 2 µl
- 5× Green GoTaq Flexi Buffer, 5 µl
- GoTaq Flexi DNA Polymerase, 0.125 µl
- Ultrapure ddH2O, 11.375 µl
Screening of six to eight colonies per construct is recommended. If a negative control ligation was performed during step 7, this can be used to estimate the level of background due to self-ligated vector. If this is really low, screening of two to four colonies should be sufficient. Since the efficiency of shRNA cloning for most sequences is very high (>90%), colony PCR screening might be entirely bypassed (skip steps 10 to 15). If this is the case, it is recommended to isolate six to eight clones for plasmid miniprep and confirm successful cloning by BglII digest followed by DNA sequencing (steps 16 to 18).
11. For each clone to be analyzed, prepare two sets of sterile PCR tubes containing 200 µl of LB broth with 100 µg/ml of ampicillin,and 10 µl of ultrapure ddH2O, respectively (use a 96 well plate if analyzing a large number of clones).
12. Using a micro-pipette set on 5 µl and a sterile tip with an aerosol-barrier filter, pick a single bacterial colony from the plate from step 9. Pipet up and down five times into the tube with ddH2O to break down the colony, transfer 5 µl of the bacterial suspension to the PCR tube, and pipet the remaining 5 µl to the tube with LB broth. Change tip and repeat this step for all clones to be analyzed. At the end of this step transfer the tubes with LB broth to 4°C storage for later use.
13. To the tube prepared for the negative control, add 5 µl of uncut pAAV-Puro_siKD plasmid diluted to 0.2ng/µl. This reaction will facilitate determining positive clones based on the size of the PCR band (step 15).
14. Perform the PCR according to the following protocol:
- 95°C, 5 min
- 95°C, 30 sec
- 60°C, 30 sec
- 72°C, 1 min
- Repeat steps 2 to 4, 34 times:
- 72°C, 2 min
- 10°C hold
15. Run 7 µl of the PCR reaction on a 1.5% (w/v) agarose gel containing 0.5 μg/ml ethidium bromide. Run at 100 V for at least 30 min, image the gel using an UV transilluminator, and identify the clones giving a product which size is approximately 350 bp (depending on the size of the shRNA sequence; Figure 4D).
Self-ligated vectors will result into a product of 295 bp, which can be identified from the negative control from step 13 (Figure 4D). If the size difference between the shRNA containing plasmids and the empty control vector is unclear, run the gel for a longer time until the size resolution is sufficient.
16. Inoculate the bacterial clones containing the correct ligation product using the liquid culture from step 12 by transferring 100 µl of the bacterial suspension to a microbiology tube containing 4 ml of LB broth with 100 µg/ml of ampicillin. Incubate in a bacterial culture orbital shaker at 37°C overnight (16 hr) while shaking at 225 rpm.
We recommend expanding two to four clones in order to have some backup should the subsequent sequencing results demonstrate any mutation in the shRNA sequence (step 19). Store the bacterial plate from step 9 at 4°C for up to 2 weeks. This can be reused to repeat steps 12 to 15 if required.
2G: Isolation and quality control of the plasmid
17. Isolate plasmids from bacterial cultures (step 16) using the QIAprep Spin Miniprep Kit following manufacturer’s instructions. Save 200 µl of the bacterial culture at 4°C.
18. Optional: Confirm shRNA cloning by performing a diagnostic restriction digestion with BglII using pAAV-Puro_siKD as control.
Correct recombinants containing the shRNA will not be linearized by BglII (provided no new BglII site is found in the shRNA sequence), as this restriction site is destroyed during the cloning. If the plasmids have been pre-screened by colony PCR (steps 10 to 15), this step is redundant and can be omitted.
19. Perform Sanger DNA sequencing using the siKD_fw primer to confirm the presence of the shRNA and its correct sequence.
The strong hairpin secondary structure of certain shRNAs can lead to difficulties in Sanger sequencing. In this case, try sequencing with siKD_rev primer and ask the sequencing company to use conditions appropriate for sequencing of GC-rich templates or sequences with a strong secondary structure. We recommend using the plasmid map generated in step 7 of Basic Protocol 1 as reference to align the sequencing results.
20. Using the liquid culture from step 17, expand the bacterial clones containing the correct shRNA in 50 ml of LB broth with 100 µg/ml of ampicillin. Incubate in a bacterial culture orbital shaker at 37°C overnight (16 hr) while shaking at 225 rpm.
The liquid culture from step 17 should be used within a week of storing at 4°C. Otherwise we recommend re-transforming the plasmid from step 17, picking a fresh bacterial colony, pre-inoculating it in 2 ml of LB broth with 100 µg/ml of ampicillin for 6 hr, and then diluting it 1:1000 in 50 ml of LB broth to be incubated as described above.
21. Prepare a glycerol stock to be stored at –80°C for long-term back up by mixing 200µl of sterile autoclaved 50%(v/v) glycerol in ddH2O with 200 µl of the bacterial culture from step 20.
Glycerol stocks can be stored for up to 10 years and used to recover the plasmid. For this, streak the glycerol stock as in step 9 then repeat step 20.
22. Use the remaining culture to isolate the plasmid using the QIAfilter Midiprep Kit. Resuspend the purified plasmid in elution buffer (10 mM Tris·Cl, pH 7.5 in ddH2O) at a concentration of 500 to 1000 ng/µl and store at –20°C for up to a year.
Avoid repeated freeze-thaw cycles of the plasmid. ddH2O can be used instead of elution buffer, but the shelf life of the plasmids will decrease. We did not notice interference of elution buffer with the subsequent gene targeting steps. We advise using freshly prepared plasmids (less than a month of storage) to increase the efficiency of gene targeting. We strongly advise repeating step 19 to confirm the shRNA sequence before proceeding with gene-targeting experiments.
Basic Protocol 3: Generation of OPTiKD hPSCs
In this third part of the procedure, the targeting vector with the inducible shRNA(s) is integrated into the AAVS1 locus to generate OPTiKD hPSCs (Figure 5A). First, hPSCs are co-transfected with the targeting vector and two AAVS1 ZFN plasmids. Correctly gene-targeted hPSCs are then selected by addition of puromycin to eliminate untargeted cells. The resulting OPTiKD cells can be used for experimental purposes either directly as a pool arising from various targeting events or after clonal selection of individual sublines (Support Protocol 2).
In this third part of the procedure, the targeting vector with the inducible shRNA(s) is integrated into the AAVS1 locus to generate OPTiKD hPSCs (Figure 5A). First, hPSCs are co-transfected with the targeting vector and two AAVS1 ZFN plasmids. Correctly gene-targeted hPSCs are then selected by addition of puromycin to eliminate untargeted cells. The resulting OPTiKD cells can be used for experimental purposes either directly as a pool arising from various targeting events or after clonal selection of individual sublines (Support Protocol 2).
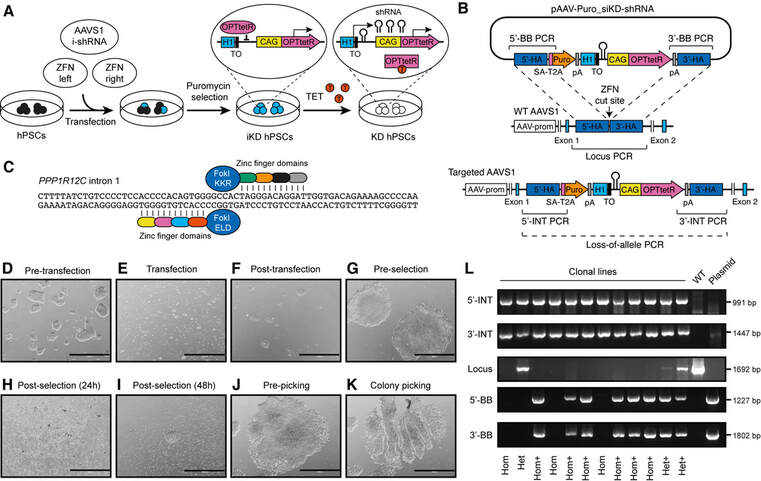
Figure 5. Generation of OPTiKD hPSCs. (A) Schematic of the gene targeting procedure described in Basic Protocol 3. The resulting OPTiKD transgenic allele and its functionality in presence or absence of tetracycline (TET) are shown. ZFN: zinc finger nuclease; i-shRNA: inducible shRNA; iKD: inducible knockdown; KD: knockdown. (B) Schematic of the AAVS1 gene-targeting event that generates the OPTiKD transgenic allele through homologousre- combination of the donor plasmid. The genotyping strategies used to identify correctly targeted hPSCs are shown (Support Protocol 2). Locus PCR: PCR product of wild-type AAVS1 locus (indicating a non-targeted allele); Loss-of- allele: potential PCR amplification that fails onto the targeted allele due to large size and high GC-content; 5'/3'-INT PCR: PCR product of transgene 5'/3'-end integration region (indicative of expected transgene targeting); 5'/3'-BB PCR: PCR product of vector backbone 5'/3'-end (indicative of nonspecific off-target plasmid integration). (C) Location of the binding sites for the AAVS1 ZFN. FokI ELD/KKR: mutant obligate heterodimer FokI endonuclease domains (Section 3.A; see Basic Protocol 3 introduction). (D-K) Representative phase-contrast images of hPSCs at the indicated stages of the gene targeting procedure. Scale bars: 1000 μm. (L) Representative example of genotyping results from 12 screened OPTiKD clonal sublines (refer to panel B). The inferred genotypes are indicated. Hom: homozygous; Het: heterozygous; +: off-target plasmid integration. WT: control PCR from wild-type hPSCs; Plasmid: positive control PCR.
3.A: Gene targeting of the AAVS1 locus
As introduced in Section 2.A (see introduction to Basic Protocol 2), selective integration of the targeting vector into the AAVS1 locus is facilitated by ZFNs, which induce site-specific double-strand breaks that are then repaired by HDR using the targeting vector as template (Figure 5B). This leads to integration of the transgenic cassette placed between the two homology arms, which contains all functional elements of the OPTiKD method. The ZFN-based gene targeting procedure was originally described by the Jaenisch laboratory (Hockemeyer et al., 2009), which also developed a similar approach based on Transcription Activator-Like Effector Nucleases (TALENS; Hockemeyer et al., 2011). More recently, analogous strategies based on Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 gene editing have been also reported (Mali et al., 2013; Oceguera-Yanez et al., 2016; Sim, Cardenas-Diaz, French, & Gadue, 2015). In all cases, the engineered nucleases recognize a very similar (if not identical) sequence between exons 1 and 2 of the PPP1R12C gene on human chromosome 19.
Our protocol relies on a modified version of the original ZFN-based method by Jaenisch and colleagues (Hockemeyer et al., 2009). ZFNs consist of two functional domains: a DNA-binding domain, which is classically constituted by two or more two-finger modules each recognizing a hexamer sequence, and a DNA-cleaving domain comprising the nuclease domain of FokI (Gaj et al., 2013). Such a domain must dimerize in order to cleave the DNA, and thus a pair of adjacent ZFNs each targeting opposite DNA strands are required in order to induce a double-strand break (Figure 5C). In our method, the original ZFN plasmids have been modified in the following ways: (1) the two FokI domains carry respectively the ELD (Q486E, I449L, and N496D) and KKR (E490K, I538K, and H537R) mutations, which provide superior cleavage activity while minimizing non specific cleavage by suppressing homodimerization (Doyonetal.,2011); (2) the ZFN cDNAs were codon-optimized for mammalian expression, which increases the steady-state protein expression; (3) the ZFNs are expressed under the control of the strong cytomegalovirus (CMV) promoter, which ensures rapid and transient expression upon transfection. The resulting plasmids (pZFN_AAVS1-L-ELD and pZFN_AAVS1R-KKR) are approximately 4 kbp long, and should be expanded in transformed bacteria cultured in presence of 50 µg/ml kanamycin. These recommended plasmids are available upon request from the corresponding authors. The plasmid repository Addgene offers alternative ZFNs (#60915 and #60916; Sim et al., 2015) and TALENs (#59025 and #59026; Gonz´alez et al., 2014) that are compatible with our targeting vector, as they induce double-strand breaks in the same genomic region. Plasmids for CRISPR/Cas9 editing of the AAVS1 locus are also available from Addgene, but in this case modifications of the targeting vector might be required to ensure that these will not be cut by the Cas9 protein (for instance by mutating the protospacer adjacent motif sequence). If using nucleases different from the ones we recommend, the gene editing procedure might need to be re-optimized, in particular the ratio of nuclease plasmids to targeting vector.
3.B: Plasmids delivery and selection of gene targeted lines
The two AAVS1 ZFN plasmids and the targeting vector must be co-expressed to initiate gene targeting (Figure 5A). For this, we recommend the use of GeneJuice, a non-lipid-based chemical transfection reagent which is minimally toxic to hPSCs (Figure 5F). With the cost-effective and simple method described in the following protocol we routinely achieve 20% to 40% transfection efficiency with >80% survival in a number of hPSC lines. Alternatively, we have also successfully used cationic-lipid based transfection reagents such as Lipofectamine 2000 (Bertero et al., 2016), albeit the toxicity of this method proved significant in certain hPSC lines. Finally, nucleofection represents another valid option that allows higher efficiency of plasmid delivery (up to 70%; Bertero et al., 2016) at the expense of decreased cell survival (50% or less), need for specialized equipment, and increased costs. Therefore, we only recommend nucleofection as a backup approach to improve plasmid delivery should initial transfection experiments prove inefficient in a certain hPSC line. Regardless of the method of preference, it is of crucial importance to achieve plasmid delivery in at least 20% of the cells in order to maximize the efficiency of gene targeting.
Following transfection, hPSCs are grown for a few days to allow successfully targeted cells to form a small sub-colony of 8 to 64 cells, as this will promote their survival during the subsequent drug selection step (Figure 5G). Gene-targeted hPSCs are then selected by adding puromycin to the culture medium. As described in Section 2.A (see introduction to Basic Protocol 2), the OPTiKD transgenic cassette includes a gene trap–based puromycin resistance gene (PAT) whose expression is driven by the endogenous promoter found in the AAVS1 locus (PPP1R12C gene). In our experience, the dosage of puromycin required to complete selection while inducing minimal toxicity in gene-targeted cells is similar across different hPSC lines (0.4 to 1.2 µg/ml). However, we recommend testing this aspect in each new line by performing a drug-sensitivity experiment. In addition to puromycin, the ROCK inhibitor Y-27632 can be added during the first 2 days of selection in order to promote survival of the small colonies of gene targeted cells (Watanabe et al., 2007). The vast majority of hPSCs will die following puromycin selection, and a number of small colonies should appear and gradually grow over the subsequent days of culture (Figure 5H-J). Since the efficiency of targeting using this method is extremely high, with more than 95% of clonally isolated lines showing the expected transgene in the AAVS1 locus (Bertero et al., 2016), the resulting OPTiKD hPSCs can be used directly for experimental purposes. Alternatively, individual clonal sub-lines can be isolated to generate a homogeneous population (Figure 5K), an optional procedure described in Support Protocol 2. Once drug selection is complete, addition of puromycin to the medium is not required, and depends upon user preference. Indeed, we have not observed silencing of the OPTiKD transgene even upon prolonged culture for up to 20 passages.
3.C: Culture conditions
The following protocol was optimized for hPSCs maintained in Essential 8 (E8), a commonly used and commercially available medium for hPSC culture in feeder-free, chemically defined, and xeno-free conditions (Chen et al., 2011). The protocol further relies upon seeding hPSCs on tissue culture dishes pre-coated with recombinant human Laminin-521, a physiological, chemically defined, and xeno-free substrate that supports optimal attachment and growth of genetically stable hPSCs (Luetal., 2014; Rodinetal., 2014). In our experience, these conditions maximize both the speed and efficiency when generating OPTiKD cells. We have also successfully generated feeder-free OPTiKD hPSCs in cells maintained in a chemically defined media containing bovine serum albumin (CDM-BSA) or polyvinyl acid (CDM-PVA) and seeded upon culture dishes coated with FBS and gelatin (Bertero et al., 2016; Vallier, 2011). Thus, we anticipate that the procedure described below will be applicable in a wide range of feeder-free hPSC culture conditions following potential minor optimizations. Should feeder-dependent culture of hPSCs be preferable to the user, more extensive modifications to the protocol might be required, including the use of puromycin resistant mouse embryonic fibroblasts (MEFs) such as DR4 MEFs.
The protocol below describes the procedure to generate OPTiKD hPSCs from cells cultured and transfected in a 6-well plate format. Successful transfection of a single well of 6-well plate should result in at least five colonies following drug selection (range of 5 to 100 depending on the cell line). However, when possible, we recommend transfecting two to three wells for each inducible shRNA in order to maximize the chances of success. In this case, the different wells can serve as biological replicates during subsequent experiments. If clonal isolation of OPTiKD sub-lines is preferred, having multiple wells each containing a few (5 to 20) clearly distinguishable colonies facilitates picking while minimizing potential for multiclonality in any given colony. Nevertheless, the protocol can be scaled to different plate formats (both larger and smaller) with minimal optimization. For instance, we have successfully scaled down volumes and plasmid quantities in the procedure described below to generate OPTiKD hPSCs in a 12-well plate format, which simplifies the generation of multiple OPTiKD lines in parallel.
Materials:
- Human pluripotent stem cells (hPSCs)
- Complete Essential 8 (E8) medium (see recipe)
- Opti-MEM reduced serum medium with GlutaMAX (ThermoFisher, cat. no. 51985034)
- Dulbecco’s phosphate-buffered saline (DPBS) without calcium and magnesium (ThermoFisher, cat. no. 14190250)
- Versene solution (ThermoFisher, cat. no. 15040066)
- 10 mM Y-27632 stock solution (see recipe)
- 0.4% trypan blue (ThermoFisher, cat. no. 15250061)
- GeneJuice transfection reagent (EMD Millipore, cat. no. 70967)
- pZFN_AAVS1-R-KKR [contact us, unfortunately these plasmids are not yet available on Addgene]
- pZFN_AAVS1-L-ELD [contact us, unfortunately these plasmids are not yet available on Addgene]
- pAAV-Puro_siKD-shRNA or pAAV-Puro_MsiKD-shRNAs (Basic Protocol 2, Alternate Protocol, or Support Protocol 1)
- AAVS1-CAGGS-EGFP (Addgene, cat. no. 22212)
- 10 mg/ml puromycin dihydrochloride (Sigma Aldrich, cat. no. P9620-10ML)
- Recombinant human Laminin-521-coated 6-well culture plates (see recipe)
- 15-ml conical centrifuge tubes
- Inverted microscope equipped with fluorescent lamp and filters for FITC (EGFP) detection
- Hemacytometer
Additional reagents and equipments for culture of human pluripotent stem cells (Costa et al., 2008)
3.D: Preparatory steps
1. Grow hPSCs to 50% to 60% confluence in complete E8 medium without antibiotics on 6-well plates coated with Laminin-521 (Figure 5D). Maintain cells in an incubator with 5% CO2 and 95% humidity.
Cells to be used for transfection should be subconfluent and within their exponential growth phase for optimal transfection. Estimate the necessary cell amount based on the number of wells to be transfected (300,000 cells per well; step 8). Antibiotics are toxic during transfection due to the increased cell membrane permeability. Therefore, it is recommended to omit them from the culture medium for at least 2 days both before and after transfection.
2. On the day before setting up the transfection, pre-coat overnight new 6-well plates with Laminin-521 as described in Reagents and Solutions.
We recommend preparing two to three wells for each inducible shRNA targeting vector to be transfected (Section 3.C; see introduction to Basic Protocol 3). A mock transfection without any plasmids is recommended as a negative control to monitor the efficiency of drug selection (step 13). Finally, a transfection using AAVS1-CAGGS-EGFP (to be used instead of pAAV-Puro_siKD-shRNA) is also recommended as positive control, as it will allow monitoring transfection and gene targeting efficiency by visual inspection of EGFP fluorescence (steps 11 to 13). For both these controls, a single well is sufficient.
3. Pre-warm the new Laminin-521 coated plates in the incubator, allow Opti-MEM and E8 to reach room temperature, thaw and gently mix the plasmids and Y-27632, and pre-warm Versene at 37°C in a heated water bath.
3.E: Collection of hPSCs
4. Aspirate hPSC culture medium, rinse the cells with 2 ml of DPBS without calcium and magnesium, and incubate in 2 ml of Versene for 3 to 6 min at 37°C in the incubator until colonies are ready for mechanical passaging.
Volumes are for one well of 6-well plate. The EDTA contained in Versene chelates divalent ions such as calcium and magnesium, thus inhibiting cell-cell and cell-matrix contacts. The timing of Versene incubation is dependent on the cell line and growth conditions and must be adjusted empirically. When monitored under the microscope, the edges of hPSC colonies must be lifting while cells at the center of the colonies must be rounding up. However, do not wait until the whole colony is lifting.
5. Gently aspirate Versene and mechanically lift cells by adding 1 ml of complete E8 supplemented with 10 µM Y-27632 (E8+Y). Gently pipet up and down five to ten times to mechanically triturate the colonies and transfer to a 15-ml conical tube. Wash the well once with 1 ml of E8+Y to collect residual cells and transfer to the same conical tube.
Before lifting the cells, confirm that the colonies are readily detaching from the plate by gently tapping it on the side. If necessary, add 100 µl of Versene and incubate the cells at room temperature for up to 5 min. When lifting the cells, add medium gradually and proceed from the bottom to the top of the well to prevent the calcium found in E8 from inactivating Versene in cells that have not been yet mechanically detached.
6. Inspect an aliquot of the cell suspension under the microscope to confirm dissociation to near single cells. If necessary, triturate again by gently pipetting 5 to 10 times.
Aim to generate a suspension with approximately 50% single cells while the rest are found in small clumps of 2 to 10 cells (Figure 5E). Do not over-triturate and minimize mechanical stress on the cells. 7. Dilute 50 µl of cell suspension in an equal volume of trypan blue solution and perform live cell count using a hemacytometer. Dilute the cell suspension to 150,000 live cells/ml in E8+Y. hPSC viability should be >95%. Prepare sufficient volume for the number of wells to be plated (plus some extra to account for pipetting errors); 300,000 cells are required for each condition.
3.F: Transfection of hPSCs
8. Remove the excess Lamin-521 coating solution from pre-coated 6-well plates (step 1) and immediately aliquot 300,000 hPSCs per well in a volume of 2 ml. Gently transfer the plate to the incubator.
Do not let the coated surface dehydrate, as this will inactivate the Laminin-521 coating. When plating the cells, avoid creating turbulences that can lead to uneven plating by gently pipetting the cell suspension. Do not shake the plate to prevent cell clumping in the middle of the well.
9. For each well to be transfected, prepare the transfection mix according to the following steps:
- Add 6 µl of GeneJuice to 100 µl of Opti-MEM in a microcentrifuge tube.
- Immediately mix by vortexing at maximum speed for 3 sec.
- Incubate at room temperature for 5 min.
- Add the following plasmids:
- pZFN_AAVS1-R-KKR, 667 ng
- pZFN_AAVS1-L-ELD, 667 ng
- pAAV-Puro_siKD-shRNA, 667 ng
- Immediately mix by pipetting 3 to 5 times. Do not vortex
- Incubate at room temperature for 15 min.
10. Pipet the transfection mix drop-by-drop onto the hPSCs from step 8. Cover all the well surface by following a spiral pattern from the outside to the inside to ensure homogeneous dispersion. Transfer the plate to the incubator and gently rock back and forth, left and right two to three times to further distribute the transfection mix. Incubate overnight (16 to 20 hr).
hPSCs readily adhere to Laminin-521 and should be firmly attached to the bottom of the plate at this stage.
3.G: Selection of gene targeted hPSCs
11. On the day following transfection, aspirate the culture medium, gently rinse the cells once with 2 ml of DPBS without calcium and magnesium, and add 2 ml of fresh complete E8 medium.
From here onwards, the culture medium must be replaced daily without any cell wash unless stated otherwise. Inspect the cells to confirm lack of bacterial contamination. There should be very little toxicity due to the transfection reagent, with most of the cells being attached and forming small colonies (Figure 5F). hPSCs might look spindle-shaped at this stage, but the morphology will improve following removal of ROCK inhibitor from the medium. If a positive control transfection with AAV-CAGGS-EGFP has been performed (step 9), EGFP fluorescence should be readily detectable in at least 20% of the cells.
12. After approximately 48 hr (on the third day following transfection), begin selection by adding 1 µg/ml puromycin to the culture medium.
Selection should be started when the cells reach approximately 50% to 70% confluence [Section 3.B (see introduction to Basic Protocol 3); Figure 5G]. If a positive-control transfection with AAV-CAGGS-EGFP has been performed (step 9), EGFP-positive clusters of 8 to 32 cells should be visible. The optimal dose of puromycin should be determined for every new cell line (Section 3.B; see introduction to Basic Protocol 3). Puromycin should be added to the medium from now on except when passaging the colonies (step 14), in order to avoid excessive stress on single cells. Once the OPTiKD hPSC line has been established and banked puromycin can be withdrawn (Section 3.B; see introduction to Basic Protocol 3). Addition of 10 µM Y-27632 (ROCK inhibitor) during the first 48 hr of selection can increase survival of gene targeted colonies.
13. After approximately 48 hr (on the fifth day following transfection) selection should be completed (Figure 5H-I).
All cells should be dead in the negative control transfection without the targeting plasmid (step 9). If a positive control transfection with AAV-CAGGS-EGFP has been performed (step 9), colonies with homogeneous EGFP expression should be visible.
14. After approximately 2 to 5 days (on the seventh to tenth day following transfection), individual hPSC colonies will reach an appropriate size for passaging (500 to 1000 µm; Figure 5J). The resulting OPTiKD hPSCs can be directly used for validation and experimental purposes (Basic Protocol 4). Alternatively, they can first be clonally isolated (Support Protocol 2).
The growth rate will be cell-line dependent and certain hPSCs might require up to 20 days following transfection to reach this stage. If multiple wells have been transfected with the same inducible shRNA vector, we advise keeping them separate as biological replicates. The OPTiKD line should be expanded and banked for long-term storage in liquid nitrogen as soon as possible. Furthermore, it is recommended to perform standard quality controls such as karyotyping and testing to exclude mycoplasma contamination.
Basic protocol 4 - Validation of OPTiKD hPSCs
In this last section we describe the validation of OPTiKD hPSCs. Given that this aspect will vary broadly depending on the gene of interest, the reagents that are available to study such gene, and the experimental question being investigated, a conventional step-by-step protocol would be poorly suited to this task. Therefore, we instead present general considerations in a discursive form to help the user design the most appropriate validation experiments.
4.A – Inducible knockdown using OPTiKD hPSCs
Knockdown can be induced by simple addition of the drug tetracycline the culture media at a recommended concentration of 1 µg/ml, a dose that does not induce toxicity to hPSCs and does not interfere with hPSC differentiation. Note that the tetracycline analogue doxycycline is equally effective to induce knockdown in hPSCs, but we have not extensively tested potential side effects during hPSC differentiation. Tetracycline is a light-sensitive reagent and should be protected from direct illumination during preparation, storage, and usage. Aqueous solutions of tetracycline are unstable, and they gradually become turbid due to hydrolysis and precipitation. Thus, aqueous tetracycline solutions should be stored as single-use aliquots at -80 °C (see Reagents and Solutions). Once diluted in culture media and maintained at 37 °C, the half-life of tetracycline is approximately 24 hours. Therefore, we recommend performing media changes at least every other day. It is best to add fresh tetracycline to the culture media just before use, but should it be preferable to prepare larger volumes of medium these should be stored at 4 °C and used within a week of preparation.
It is very important to keep in mind that tetracycline is a widely used antibiotic in livestock animals such as cows and horses. Therefore animal-derived products such as fetal bovine serum (FBS) and bovine serum albumin (BSA) carry the risk of being contaminated with tetracycline. It is therefore important to batch-test animal-derived reagents (in particular FBS and derivatives) for lack of detectable tetracycline contamination before use in OPTiKD hPSCs. Wherever possible, we recommend substituting such reagents with tetracycline-free validated alternatives that are available from commercial suppliers.
4.B – Validation of OPTiKD hPSCs
We recommend validating newly generated OPTiKD hPSCs both at the transcript and protein level. Importantly, if expression of the target gene is low or absent in hPSCs, the cells should be first differentiated into an appropriate cell lineage expressing such gene. This will usually be the cell type to be functionally analyzed in follow up experiments, but should differentiation into such lineage be too expensive, laborious, and/or time consuming, validation can be instead performed in any alternative hPSC-derived cell type expressing similar levels of the gene. Transcript levels can be readily monitored by quantitative real-time PCR (qPCR), which is our preferred validation method given its simplicity, cost-effectiveness, and wide applicability to any gene of interest. When using the ΔΔCt approach to quantify relative mRNA expression levels over control samples (Livak and Schmittgen, 2001), it is important to validate that the expression of the chosen housekeeping gene is not sensitive to the addition of tetracycline. Validation by qPCR can be performed after 24 to 48 hours following induction of knockdown.
Once transcript-level knockdown has been confirmed, it is important to validate loss of the protein product. This relies on the availability of a good antibody against the protein of interest and is therefore not always possible. If such an antibody exists, we recommend to initially validate OPTiKD cells by Western blot which allows for relatively simple and rapid semi-quantitative evaluation of protein knockdown. Flow-cytometry can provide and alternative and more quantitative assessment with the added advantage that this is evaluated at the single cell level and thus interrogates the heterogeneity in the population. For example, if a pseudo-clonal OPTiKD subline has been selected (Support Protocol 2) flow-cytometry can confirm that the knockdown is homogeneous and that the selected subline is most likely clonal. If OPTiKD cells have not been clonally isolated, it is important to ensure that the majority of the population (>90%) undergoes inducible knockdown before proceeding with extensive follow-up experiments. Immunocytochemistry is an alternative way to assess population heterogeneity in OPTiKD cells, while it also allows for spatial and morphological analyses. However, immunocytochemistry is poorly quantitative and prone to false-positive signals, thus usually requiring extensive optimization. Regardless of the technique used, wherever possible it is important that protein-level validation is performed while including appropriate controls, one of the most important being a cell type not expressing the protein of interest. This type of validation is better performed after prolonged tetracycline treatment of at least 5 to 10 days in order to ensure evaluation of the maximal level of protein knockdown that can be achieved.
11. On the day following transfection, aspirate the culture medium, gently rinse the cells once with 2 ml of DPBS without calcium and magnesium, and add 2 ml of fresh complete E8 medium.
From here onwards, the culture medium must be replaced daily without any cell wash unless stated otherwise. Inspect the cells to confirm lack of bacterial contamination. There should be very little toxicity due to the transfection reagent, with most of the cells being attached and forming small colonies (Figure 5F). hPSCs might look spindle-shaped at this stage, but the morphology will improve following removal of ROCK inhibitor from the medium. If a positive control transfection with AAV-CAGGS-EGFP has been performed (step 9), EGFP fluorescence should be readily detectable in at least 20% of the cells.
12. After approximately 48 hr (on the third day following transfection), begin selection by adding 1 µg/ml puromycin to the culture medium.
Selection should be started when the cells reach approximately 50% to 70% confluence [Section 3.B (see introduction to Basic Protocol 3); Figure 5G]. If a positive-control transfection with AAV-CAGGS-EGFP has been performed (step 9), EGFP-positive clusters of 8 to 32 cells should be visible. The optimal dose of puromycin should be determined for every new cell line (Section 3.B; see introduction to Basic Protocol 3). Puromycin should be added to the medium from now on except when passaging the colonies (step 14), in order to avoid excessive stress on single cells. Once the OPTiKD hPSC line has been established and banked puromycin can be withdrawn (Section 3.B; see introduction to Basic Protocol 3). Addition of 10 µM Y-27632 (ROCK inhibitor) during the first 48 hr of selection can increase survival of gene targeted colonies.
13. After approximately 48 hr (on the fifth day following transfection) selection should be completed (Figure 5H-I).
All cells should be dead in the negative control transfection without the targeting plasmid (step 9). If a positive control transfection with AAV-CAGGS-EGFP has been performed (step 9), colonies with homogeneous EGFP expression should be visible.
14. After approximately 2 to 5 days (on the seventh to tenth day following transfection), individual hPSC colonies will reach an appropriate size for passaging (500 to 1000 µm; Figure 5J). The resulting OPTiKD hPSCs can be directly used for validation and experimental purposes (Basic Protocol 4). Alternatively, they can first be clonally isolated (Support Protocol 2).
The growth rate will be cell-line dependent and certain hPSCs might require up to 20 days following transfection to reach this stage. If multiple wells have been transfected with the same inducible shRNA vector, we advise keeping them separate as biological replicates. The OPTiKD line should be expanded and banked for long-term storage in liquid nitrogen as soon as possible. Furthermore, it is recommended to perform standard quality controls such as karyotyping and testing to exclude mycoplasma contamination.
Basic protocol 4 - Validation of OPTiKD hPSCs
In this last section we describe the validation of OPTiKD hPSCs. Given that this aspect will vary broadly depending on the gene of interest, the reagents that are available to study such gene, and the experimental question being investigated, a conventional step-by-step protocol would be poorly suited to this task. Therefore, we instead present general considerations in a discursive form to help the user design the most appropriate validation experiments.
4.A – Inducible knockdown using OPTiKD hPSCs
Knockdown can be induced by simple addition of the drug tetracycline the culture media at a recommended concentration of 1 µg/ml, a dose that does not induce toxicity to hPSCs and does not interfere with hPSC differentiation. Note that the tetracycline analogue doxycycline is equally effective to induce knockdown in hPSCs, but we have not extensively tested potential side effects during hPSC differentiation. Tetracycline is a light-sensitive reagent and should be protected from direct illumination during preparation, storage, and usage. Aqueous solutions of tetracycline are unstable, and they gradually become turbid due to hydrolysis and precipitation. Thus, aqueous tetracycline solutions should be stored as single-use aliquots at -80 °C (see Reagents and Solutions). Once diluted in culture media and maintained at 37 °C, the half-life of tetracycline is approximately 24 hours. Therefore, we recommend performing media changes at least every other day. It is best to add fresh tetracycline to the culture media just before use, but should it be preferable to prepare larger volumes of medium these should be stored at 4 °C and used within a week of preparation.
It is very important to keep in mind that tetracycline is a widely used antibiotic in livestock animals such as cows and horses. Therefore animal-derived products such as fetal bovine serum (FBS) and bovine serum albumin (BSA) carry the risk of being contaminated with tetracycline. It is therefore important to batch-test animal-derived reagents (in particular FBS and derivatives) for lack of detectable tetracycline contamination before use in OPTiKD hPSCs. Wherever possible, we recommend substituting such reagents with tetracycline-free validated alternatives that are available from commercial suppliers.
4.B – Validation of OPTiKD hPSCs
We recommend validating newly generated OPTiKD hPSCs both at the transcript and protein level. Importantly, if expression of the target gene is low or absent in hPSCs, the cells should be first differentiated into an appropriate cell lineage expressing such gene. This will usually be the cell type to be functionally analyzed in follow up experiments, but should differentiation into such lineage be too expensive, laborious, and/or time consuming, validation can be instead performed in any alternative hPSC-derived cell type expressing similar levels of the gene. Transcript levels can be readily monitored by quantitative real-time PCR (qPCR), which is our preferred validation method given its simplicity, cost-effectiveness, and wide applicability to any gene of interest. When using the ΔΔCt approach to quantify relative mRNA expression levels over control samples (Livak and Schmittgen, 2001), it is important to validate that the expression of the chosen housekeeping gene is not sensitive to the addition of tetracycline. Validation by qPCR can be performed after 24 to 48 hours following induction of knockdown.
Once transcript-level knockdown has been confirmed, it is important to validate loss of the protein product. This relies on the availability of a good antibody against the protein of interest and is therefore not always possible. If such an antibody exists, we recommend to initially validate OPTiKD cells by Western blot which allows for relatively simple and rapid semi-quantitative evaluation of protein knockdown. Flow-cytometry can provide and alternative and more quantitative assessment with the added advantage that this is evaluated at the single cell level and thus interrogates the heterogeneity in the population. For example, if a pseudo-clonal OPTiKD subline has been selected (Support Protocol 2) flow-cytometry can confirm that the knockdown is homogeneous and that the selected subline is most likely clonal. If OPTiKD cells have not been clonally isolated, it is important to ensure that the majority of the population (>90%) undergoes inducible knockdown before proceeding with extensive follow-up experiments. Immunocytochemistry is an alternative way to assess population heterogeneity in OPTiKD cells, while it also allows for spatial and morphological analyses. However, immunocytochemistry is poorly quantitative and prone to false-positive signals, thus usually requiring extensive optimization. Regardless of the technique used, wherever possible it is important that protein-level validation is performed while including appropriate controls, one of the most important being a cell type not expressing the protein of interest. This type of validation is better performed after prolonged tetracycline treatment of at least 5 to 10 days in order to ensure evaluation of the maximal level of protein knockdown that can be achieved.
REAGENTS AND SOLUTIONS
Use deionized, distilled water (ddH2O) or equivalent in recipes and protocol steps.
Agarose gel (1%, 1.5%, or 4%) in TBE for DNA electrophoresis Dissolve the required amount of agarose I powder [molecular biology grade (ThermoFisher, cat. no. 17850) or UltraPure Low Melting Point Agarose (ThermoFisher, cat. no. 16520050) as called for in protocol] in 1×TBE buffer (see recipe) in a glass bottle. Place a cap on the bottle but leave loose. Incubate at room temperature for 15 min to pre-dissolve. Microwave for about 1 to 2 min or until all of the powder is fully dissolved, but do not let the solution boil. Allow the solution to cool at room temperature for 5 to 10 min (the temperature of the solution should not go below 65°C to prevent premature gelling) and add 10 mg/ml ethidium bromide (Sigma-Aldrich, cat. no. E1510-10ML) to a final concentration of 0.5 µg/ml. After mixing, pour the solution into a gel casting tray equipped with the appropriate combs and let the gel set for 15 to 30 min before use.
Also see Voytas (2001).
Essential 8 (E8) medium, complete
Add 10 ml of provided 50x Essential 8 supplement to a 500-ml bottle of Essential 8 medium (ThermoFisher, A1517001) and mix by gently inverting (avoid making bubbles, to prevent denaturation of the cytokines). Optionally, add 100x penicillin streptomycin (10,000 U/ml; ThermoFisher, 15140122) to a 1x concentration. Store complete Essential 8 medium at 4°C and use within 7 days to prevent loss of potency of the associated cytokines.
Laminin-521-coated culture plates
Thaw Laminin-521 stock solution (BioLamina, cat.no. LN521-02) at 4°C overnight and mix well by inversion before use. To prepare 1 ml of coating solution for final coating at approximately 0.5 µg/cm2, add 50 µl of the stock to 950 µl of DPBS with calcium and magnesium (ThermoFisher, cat. no. 14040133). Add 1 ml to one well of 6-well plate (Corning, cat. no. 3516) or 0.5 ml to one well of a 12-well plate (Corning, cat. no. 3513). Swirl to ensure that the bottom surface is entirely covered. Tightly seal and incubate at 4°C overnight. Alternatively, plates can be coated for 2 hr at 37 °C. Pre-coated plates can be stored at 4°C for up to 2 weeks provided they do not dry out.
LB agar plates
Dissolve 20 g of agar powder (e.g., BD Difco) in 1 liter of LB broth and mix well. Autoclave on liquid cycle and let cool until it is warm enough to touch (approximately 50°C). Add the appropriate antibiotic [ampicillin (Sigma-Aldrich, cat. no. A535410ML)] to 100 µg/ml and swirl to mix (do not shake as this will create bubbles). Pour into 10-cm petri dishes to completely cover the bottom surface. Allow the plates to set for 2 hr at room temperature and store at 4°C up to 3 months.
Luria Bertani (LB) broth
To prepare a 1-liter solution add:
Tetracycline stock solution
Prepare this reagent protected from direct illumination. Dissolve 50 mg of tetracycline hydrochloride in 5 ml of ultrapure ddH2O. The resulting solution should have a mild yellow-orange color. Filter-sterilize using a 0.22 µm filter and prepare single-use 5 or 10 µl aliquots. Store at -80 °C for up to 6 months.
Tris-borate-EDTA (TBE) buffer
To prepare a 1-liter 5x stock solution add:
Y-27632 stock solution, 10 mM stock
To obtain a 10 mM solution dissolve 1 mg of Y-27632 powder (Tocris, cat. no. 1254) into 312.25 µl of DMSO. Prepare single-use aliquots and store at –20 °C for up to 6 months.
Use deionized, distilled water (ddH2O) or equivalent in recipes and protocol steps.
Agarose gel (1%, 1.5%, or 4%) in TBE for DNA electrophoresis Dissolve the required amount of agarose I powder [molecular biology grade (ThermoFisher, cat. no. 17850) or UltraPure Low Melting Point Agarose (ThermoFisher, cat. no. 16520050) as called for in protocol] in 1×TBE buffer (see recipe) in a glass bottle. Place a cap on the bottle but leave loose. Incubate at room temperature for 15 min to pre-dissolve. Microwave for about 1 to 2 min or until all of the powder is fully dissolved, but do not let the solution boil. Allow the solution to cool at room temperature for 5 to 10 min (the temperature of the solution should not go below 65°C to prevent premature gelling) and add 10 mg/ml ethidium bromide (Sigma-Aldrich, cat. no. E1510-10ML) to a final concentration of 0.5 µg/ml. After mixing, pour the solution into a gel casting tray equipped with the appropriate combs and let the gel set for 15 to 30 min before use.
Also see Voytas (2001).
Essential 8 (E8) medium, complete
Add 10 ml of provided 50x Essential 8 supplement to a 500-ml bottle of Essential 8 medium (ThermoFisher, A1517001) and mix by gently inverting (avoid making bubbles, to prevent denaturation of the cytokines). Optionally, add 100x penicillin streptomycin (10,000 U/ml; ThermoFisher, 15140122) to a 1x concentration. Store complete Essential 8 medium at 4°C and use within 7 days to prevent loss of potency of the associated cytokines.
Laminin-521-coated culture plates
Thaw Laminin-521 stock solution (BioLamina, cat.no. LN521-02) at 4°C overnight and mix well by inversion before use. To prepare 1 ml of coating solution for final coating at approximately 0.5 µg/cm2, add 50 µl of the stock to 950 µl of DPBS with calcium and magnesium (ThermoFisher, cat. no. 14040133). Add 1 ml to one well of 6-well plate (Corning, cat. no. 3516) or 0.5 ml to one well of a 12-well plate (Corning, cat. no. 3513). Swirl to ensure that the bottom surface is entirely covered. Tightly seal and incubate at 4°C overnight. Alternatively, plates can be coated for 2 hr at 37 °C. Pre-coated plates can be stored at 4°C for up to 2 weeks provided they do not dry out.
LB agar plates
Dissolve 20 g of agar powder (e.g., BD Difco) in 1 liter of LB broth and mix well. Autoclave on liquid cycle and let cool until it is warm enough to touch (approximately 50°C). Add the appropriate antibiotic [ampicillin (Sigma-Aldrich, cat. no. A535410ML)] to 100 µg/ml and swirl to mix (do not shake as this will create bubbles). Pour into 10-cm petri dishes to completely cover the bottom surface. Allow the plates to set for 2 hr at room temperature and store at 4°C up to 3 months.
Luria Bertani (LB) broth
To prepare a 1-liter solution add:
- 950 ml of ddH2O
- 10 g Tryptone
- 10 g NaCl
- 5 g Yeast extract
Tetracycline stock solution
Prepare this reagent protected from direct illumination. Dissolve 50 mg of tetracycline hydrochloride in 5 ml of ultrapure ddH2O. The resulting solution should have a mild yellow-orange color. Filter-sterilize using a 0.22 µm filter and prepare single-use 5 or 10 µl aliquots. Store at -80 °C for up to 6 months.
Tris-borate-EDTA (TBE) buffer
To prepare a 1-liter 5x stock solution add:
- 800 ml of ddH2O
- 54 g of Tris base
- 27.5 g of boric acid
- 20 ml of 0.5 M EDTA (pH 8.0)
Y-27632 stock solution, 10 mM stock
To obtain a 10 mM solution dissolve 1 mg of Y-27632 powder (Tocris, cat. no. 1254) into 312.25 µl of DMSO. Prepare single-use aliquots and store at –20 °C for up to 6 months.
References
- Avior, Y., Sagi, I., and Benvenisty, N. (2016). Pluripotent stem cells in disease modelling and drug discovery. Nature reviews. Molecular cell biology, 17, 170–182. doi: 10.1038/nrm.2015.27 .
- Bertero, A., Pawlowski, M., Ortmann, D., Snijders, K., Yiangou, L., Cardoso de Brito, M., Brown, S., Bernard, W. G., Cooper, J. D., Giacomelli, E., et al. (2016). Optimized inducible shRNA and CRISPR/Cas9 platforms for in vitro studies of human development using hPSCs. Development, 143, .
- Boettcher, M., and McManus, M. T. (2015). Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Molecular cell, 58, 575–585. doi: 10.1016/j.molcel.2015.04.028 .
- Boudreau, R. L., Martins, I., and Davidson, B. L. (2009). Artificial MicroRNAs as siRNA Shuttles: Improved Safety as Compared to shRNAs In vitro and In vivo. Molecular Therapy, 17, 169–175. doi: 10.1038/mt.2008.231 .
- Brummelkamp, T. R., Bernards, R., and Agami, R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–3. doi: 10.1126/science.1068999 .
- Cao, F., Xie, X., Gollan, T., Zhao, L., Narsinh, K., Lee, R. J., and Wu, J. C. (2010). Comparison of gene-transfer efficiency in human embryonic stem cells. Molecular imaging and biology, 12, 15–24. doi: 10.1007/s11307-009-0236-x .
- Chatzispyrou, I. A., Held, N. M., Mouchiroud, L., Auwerx, J., and Houtkooper, R. H. (2015). Tetracycline Antibiotics Impair Mitochondrial Function and Its Experimental Use Confounds Research. Cancer Research, 75, 4446–4449. doi: 10.1158/0008-5472.CAN-15-1626 .
- Chen, G., Gulbranson, D. R., Hou, Z., Bolin, J. M., Ruotti, V., Probasco, M. D., Smuga-Otto, K., Howden, S. E., Diol, N. R., Propson, N. E., et al. (2011). Chemically defined conditions for human iPSC derivation and culture. Nature methods, 8, 424–9. doi: 10.1038/nmeth.1593 .
- Chen, Y., Cao, J., Xiong, M., Petersen, A. J., Dong, Y., Tao, Y., Huang, C. T.-L., Du, Z., and Zhang, S.-C. (2015). Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell, 17, 233–244. doi: 10.1016/j.stem.2015.06.001 .
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–23. doi: 10.1126/science.1231143 .
- Cooper, G. M., and Shendure, J. (2011). Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature reviews. Genetics, 12, 628–40. doi: 10.1038/nrg3046 .
- Costa, M., Dottori, M., Sourris, K., Jamshidi, P., Hatzistavrou, T., Davis, R., Azzola, L., Jackson, S., Lim, S. M., Pera, M., et al. (2007). A method for genetic modification of human embryonic stem cells using electroporation. Nature protocols, 2, 792–6. doi: 10.1038/nprot.2007.105 .
- Cullen, B. R. (2006). Enhancing and confirming the specificity of RNAi experiments. Nature Methods, 3, 677–681. doi: 10.1038/nmeth913 .
- DeKelver, R. C., Choi, V. M., Moehle, E. a, Paschon, D. E., Hockemeyer, D., Meijsing, S. H., Sancak, Y., Cui, X., Steine, E. J., Miller, J. C., et al. (2010). Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome research, 20, 1133–42. doi: 10.1101/gr.106773.110 .
- Doyon, Y., Vo, T. D., Mendel, M. C., Greenberg, S. G., Wang, J., Xia, D. F., Miller, J. C., Urnov, F. D., Gregory, P. D., and Holmes, M. C. (2011). Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nature methods, 8, 74–9. doi: 10.1038/nmeth.1539 .
- Drost, J., van Jaarsveld, R. H., Ponsioen, B., Zimberlin, C., van Boxtel, R., Buijs, A., Sachs, N., Overmeer, R. M., Offerhaus, G. J., Begthel, H., et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature, 521, 43–7. doi: 10.1038/nature14415 .
- Ellis, J. (2005). Silencing and variegation of gammaretrovirus and lentivirus vectors. Human gene therapy, 16, 1241–6. doi: 10.1089/hum.2005.16.1241 .
- Fakhr, E., Zare, F., and Teimoori-Toolabi, L. (2016). Precise and efficient siRNA design: a key point in competent gene silencing. Cancer Gene Therapy, 23, 73–82. doi: 10.1038/cgt.2016.4 .
- Fath, S., Bauer, A. P., Liss, M., Spriestersbach, A., Maertens, B., Hahn, P., Ludwig, C., Schäfer, F., Graf, M., and Wagner, R. (2011). Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PloS one, 6, e17596. doi: 10.1371/journal.pone.0017596 .
- Gaj, T., Gersbach, C. A., and Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology, 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004 .
- Gibson, D. G., Young, L., Chuang, R.-Y., Venter, J. C., Hutchison, C. A., and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods, 6, 343–5. doi: 10.1038/nmeth.1318 .
- González, F., Zhu, Z., Shi, Z.-D., Lelli, K., Verma, N., Li, Q. V., and Huangfu, D. (2014). An iCRISPR Platform for Rapid, Multiplexable, and Inducible Genome Editing in Human Pluripotent Stem Cells. Cell Stem Cell, 15, 215–26. doi: 10.1016/j.stem.2014.05.018 .
- Gray, D. C., Hoeflich, K. P., Peng, L., Gu, Z., Gogineni, A., Murray, L. J., Eby, M., Kljavin, N., Seshagiri, S., Cole, M. J., et al. (2007). pHUSH: a single vector system for conditional gene expression. BMC biotechnology, 7, 61. doi: 10.1186/1472-6750-7-61 .
- Grimm, D., Streetz, K. L., Jopling, C. L., Storm, T. A., Pandey, K., Davis, C. R., Marion, P., Salazar, F., and Kay, M. A. (2006). Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature, 441, 537–541. doi: 10.1038/nature04791 .
- Haenebalcke, L., Goossens, S., Naessens, M., Kruse, N., Farhang Ghahremani, M., Bartunkova, S., Haigh, K., Pieters, T., Dierickx, P., Drogat, B., et al. (2013). Efficient ROSA26-based conditional and/or inducible transgenesis using RMCE-compatible F1 hybrid mouse embryonic stem cells. Stem cell reviews, 9, 774–85. doi: 10.1007/s12015-013-9458-z .
- Henriksen, J. R., Løkke, C., Hammerø, M., Geerts, D., Versteeg, R., Flaegstad, T., and Einvik, C. (2007). Comparison of RNAi efficiency mediated by tetracycline-responsive H1 and U6 promoter variants in mammalian cell lines. Nucleic acids research, 35, e67. doi: 10.1093/nar/gkm193 .
- Herbst, F., Ball, C. R., Tuorto, F., Nowrouzi, A., Wang, W., Zavidij, O., Dieter, S. M., Fessler, S., van der Hoeven, F., Kloz, U., et al. (2012). Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Molecular therapy, 20, 1014–21. doi: 10.1038/mt.2012.46 .
- Hockemeyer, D., Soldner, F., Beard, C., Gao, Q., Mitalipova, M., DeKelver, R. C., Katibah, G. E., Amora, R., Boydston, E. a, Zeitler, B., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature biotechnology, 27, 851–7. doi: 10.1038/nbt.1562 .
- Hockemeyer, D., Wang, H., Kiani, S., Lai, C. S., Gao, Q., Cassady, J. P., Cost, G. J., Zhang, L., Santiago, Y., Miller, J. C., et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nature biotechnology, 29, 731–4. doi: 10.1038/nbt.1927 .
- Jackson, A. L., Bartz, S. R., Schelter, J., Kobayashi, S. V, Burchard, J., Mao, M., Li, B., Cavet, G., and Linsley, P. S. (2003). Expression profiling reveals off-target gene regulation by RNAi. Nature Biotechnology, 21, 635–637. doi: 10.1038/nbt831 .
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–21. doi: 10.1126/science.1225829 .
- Joung, J. K., and Sander, J. D. (2013). TALENs: a widely applicable technology for targeted genome editing. Nature reviews. Molecular cell biology, 14, 49–55. doi: 10.1038/nrm3486 .
- Kappel, S., Matthess, Y., Kaufmann, M., and Strebhardt, K. (2007). Silencing of mammalian genes by tetracycline-inducible shRNA expression. Nature protocols, 2, 3257–69. doi: 10.1038/nprot.2007.458 .
- Kim, H., and Kim, J. S. (2014). A guide to genome engineering with programmable nucleases. Nature reviews. Genetics, 15, 321–34. doi: 10.1038/nrg3686 .
- Krishnan, M., Park, J. M., Cao, F., Wang, D., Paulmurugan, R., Tseng, J. R., Gonzalgo, M. L., Gambhir, S. S., and Wu, J. C. (2006). Effects of epigenetic modulation on reporter gene expression: implications for stem cell imaging. FEBS Journal, 20, 106–8. doi: 10.1096/fj.05-4551fje .
- Kun, Q., CindyTzu-Ling, H., Hong, C., W, B. L., Yuejun, C., Jingyuan, C., Lin, Y., Cornall, S., Zhongwei, D., and Su-Chun, Z. (2014). A Simple and Efficient System for Regulating Gene Expression in Human Pluripotent Stem Cells and Derivatives. Stem cells, 32, 1230–38.
- Lambeth, L. S., and Smith, C. A. (2013). Short hairpin RNA-mediated gene silencing. Methods in Molecular Biology, 942, 205–32. doi: 10.1007/978-1-62703-119-6_12 .
- Laperle, A., Hsiao, C., Lampe, M., Mortier, J., Saha, K., Palecek, S. P., and Masters, K. S. (2015). α-5 Laminin Synthesized by Human Pluripotent Stem Cells Promotes Self-Renewal. Stem Cell Reports, 5, 195–206. doi: 10.1016/j.stemcr.2015.06.009 .
- Lian, X., Hsiao, C., Wilson, G., Zhu, K., Hazeltine, L. B., Azarin, S. M., Raval, K. K., Zhang, J., Kamp, T. J., and Palecek, S. P. (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. PNAS, 109, E1848-57. doi: 10.1073/pnas.1200250109 .
- Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–408.
- Lu, H. F., Chai, C., Lim, T. C., Leong, M. F., Lim, J. K., Gao, S., Lim, K. L., and Wan, A. C. A. (2014). A defined xeno-free and feeder-free culture system for the derivation, expansion and direct differentiation of transgene-free patient-specific induced pluripotent stem cells. Biomaterials, 35, 2816–2826. doi: 10.1016/j.biomaterials.2013.12.050 .
- Luo, C., Lü, D., Pan, J., and Long, M. (2016). Improving the Gene Transfection in Human Embryonic Stem Cells: Balancing with Cytotoxicity and Pluripotent Maintenance. ACS applied materials & interfaces, 8, 8367–75. doi: 10.1021/acsami.6b00353 .
- Ma, H., Wu, Y., Dang, Y., Choi, J.-G., Zhang, J., and Wu, H. (2014). Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Molecular therapy, 3, e161. doi: 10.1038/mtna.2014.12 .
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E., and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science, 339, 823–6. doi: 10.1126/science.1232033 .
- Mandal, P. K., Ferreira, L. M. R., Collins, R., Meissner, T. B., Boutwell, C. L., Friesen, M., Vrbanac, V., Garrison, B. S., Stortchevoi, A., Bryder, D., et al. (2014). Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells using CRISPR/Cas9. Cell Stem Cell, 15, 643–652. doi: 10.1016/j.stem.2014.10.004 .
- Mandegar, M. A., Huebsch, N., Frolov, E. B., Shin, E., Truong, A., Olvera, M. P., Chan, A. H., Miyaoka, Y., Holmes, K., Spencer, C. I., et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell, 18, 541–53. doi: 10.1016/j.stem.2016.01.022 .
- Massé, J., Piquet-Pellorce, C., Viet, J., Guerrier, D., Pellerin, I., and Deschamps, S. (2011). ZFPIP/Zfp462 is involved in P19 cell pluripotency and in their neuronal fate. Experimental cell research, 317, 1922–34. doi: 10.1016/j.yexcr.2011.04.015 .
- Miyazaki, J., Satoshi, T., Araki, K., Tashiro, F., Tominaga, A., Takatsu, K., and Yamamura, K.-I. (1989). Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene, 79, 269–277. doi: 10.1016/0378-1119(89)90209-6 .
- Moffat, J., Grueneberg, D. A., Yang, X., Kim, S. Y., Kloepfer, A. M., Hinkle, G., Piqani, B., Eisenhaure, T. M., Luo, B., Grenier, J. K., et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell, 124, 1283–98. doi: 10.1016/j.cell.2006.01.040 .
- Moullan, N., Mouchiroud, L., Wang, X., Ryu, D., Williams, E., Mottis, A., Jovaisaite, V., Frochaux, M., Quiros, P., Deplancke, B., et al. (2015). Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Reports, 10, 1681–1691. doi: 10.1016/j.celrep.2015.02.034 .
- Nakagawa, S., and Cuthill, I. C. (2007). Effect size, confidence interval and statistical significance: a practical guide for biologists. Biological reviews of the Cambridge Philosophical Society, 82, 591–605. doi: 10.1111/j.1469-185X.2007.00027.x .
- Oceguera-Yanez, F., Kim, S. Il, Matsumoto, T., Tan, G. W., Xiang, L., Hatani, T., Kondo, T., Ikeya, M., Yoshida, Y., Inoue, H., et al. (2016). Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods, 101, 43–55. doi: 10.1016/j.ymeth.2015.12.012 .
- Ordovas, L., Boon, R., Pistoni, M., Chen, Y., Wolfs, E., Guo, W., Sambathkumar, R., Bobis-Wozowicz, S., Helsen, N., Vanhove, J., et al. (2015). Efficient recombinase-mediated cassette exchange in hPSCs to study the hepatocyte lineage reveals AAVS1 locus-mediated transgene inhibition. Stem Cell Reports, 5, 918–931. doi: 10.1016/j.stemcr.2015.09.004 .
- Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U., Glimm, H., Kühlcke, K., Schilz, A., Kunkel, H., et al. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nature medicine, 12, 401–9. doi: 10.1038/nm1393 .
- Pawlowski, M., Ortmann, D., Bertero, A., Tavares, J. M., Pedersen, R. A., Vallier, L., Kotter, M. R. N., Patzke, C., Acuna, C., Covy, J., et al. (2017). Inducible and Deterministic Forward Programming of Human Pluripotent Stem Cells into Neurons, Skeletal Myocytes, and Oligodendrocytes. Stem Cell Reports, 8, 803–12. doi: 10.1016/j.stemcr.2017.02.016 .
- Pei, Y., and Tuschl, T. (2006). On the art of identifying effective and specific siRNAs. Nature Methods, 3, 670–676. doi: 10.1038/nmeth911 .
- Pourquié, O., Bruneau, B., Keller, G., and Smith, A. (2015). Looking inwards: opening a window onto human development. Development, 142, 1–2. doi: 10.1242/dev.119727 .
- Ranganathan, V., Wahlin, K., Maruotti, J., and Zack, D. J. (2014). Expansion of the CRISPR-Cas9 genome targeting space through the use of H1 promoter-expressed guide RNAs. Nature communications, 5, 4516. doi: 10.1038/ncomms5516 .
- Raya, A., Rodríguez-Pizà, I., Guenechea, G., Vassena, R., Navarro, S., Barrero, M. J., Consiglio, A., Castellà, M., Río, P., Sleep, E., et al. (2009). Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature, 460, 53–9. doi: 10.1038/nature08129 .
- Rodin, S., Antonsson, L., Niaudet, C., Simonson, O. E., Salmela, E., Hansson, E. M., Domogatskaya, A., Xiao, Z., Damdimopoulou, P., Sheikhi, M., et al. (2014). Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nature Communications, 5, 1–13. doi: 10.1038/ncomms4195 .
- Sadelain, M., Papapetrou, E. P., and Bushman, F. D. (2012). Safe harbours for the integration of new DNA in the human genome. Nature reviews. Cancer, 12, 51–8. doi: 10.1038/nrc3179 .
- Shearwin, K. E., Callen, B. P., and Egan, J. B. (2005). Transcriptional interference--a crash course. Trends in genetics, 21, 339–45. doi: 10.1016/j.tig.2005.04.009 .
- Sim, X., Cardenas-diaz, F. L., French, D. L., and Gadue, P. (2015). A Doxycycline-Inducible System for Genetic Correction of iPSC Disease Models. Methods in Molecular Biology, 1353, 13–23. doi: 10.1007/7651 .
- Smith, J. R., Maguire, S., Davis, L. a, Alexander, M., Yang, F., Chandran, S., Ffrench-Constant, C., and Pedersen, R. a (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem cells, 26, 496–504. doi: 10.1634/stemcells.2007-0039 .
- Stein, S., Ott, M. G., Schultze-Strasser, S., Jauch, A., Burwinkel, B., Kinner, A., Schmidt, M., Krämer, A., Schwäble, J., Glimm, H., et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature medicine, 16, 198–204. doi: 10.1038/nm.2088 .
- Trounson, A., and DeWitt, N. D. (2016). Pluripotent stem cells progressing to the clinic. Nature Reviews Molecular Cell Biology, 17, 194–200. doi: 10.1038/nrm.2016.10 .
- Tsuneyoshi, N., Tan, E. K., Sadasivam, A., Poobalan, Y., Sumi, T., Nakatsuji, N., Suemori, H., and Dunn, N. R. (2012). The SMAD2/3 corepressor SNON maintains pluripotency through selective repression of mesendodermal genes in human ES cells. Genes & development, 26, 2471–6. doi: 10.1101/gad.201772.112 .
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., and Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nature reviews. Genetics, 11, 636–46. doi: 10.1038/nrg2842 .
- Vallier, L. (2011). Serum-Free and Feeder-Free Culture Conditions for Human Embryonic Stem Cells. Springer protocols, 690, 57–66. doi: 10.1007/978-1-60761-962-8 .
- Vaux, D. L., Fidler, F., and Cumming, G. (2012). Replicates and repeats--what is the difference and is it significant? A brief discussion of statistics and experimental design. EMBO reports, 13, 291–6. doi: 10.1038/embor.2012.36 .
- Wang, S., Shi, Z., Liu, W., Jules, J., and Feng, X. (2006). Development and validation of vectors containing multiple siRNA expression cassettes for maximizing the efficiency of gene silencing. BMC biotechnology, 6, 50. doi: 10.1186/1472-6750-6-50 .
- Watanabe, K., Ueno, M., Kamiya, D., Nishiyama, A., Matsumura, M., Wataya, T., Takahashi, J. B., Nishikawa, S., Nishikawa, S., Muguruma, K., et al. (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature biotechnology, 25, 681–6. doi: 10.1038/nbt1310 .
- Yao, S., Sukonnik, T., Kean, T., Bharadwaj, R. R., Pasceri, P., and Ellis, J. (2004). Retrovirus silencing, variegation, extinction, and memory are controlled by a dynamic interplay of multiple epigenetic modifications. Molecular therapy, 10, 27–36. doi: 10.1016/j.ymthe.2004.04.007 .
- Zafarana, G., Avery, S. R., Avery, K., Moore, H. D., and Andrews, P. W. (2009). Specific knockdown of OCT4 in human embryonic stem cells by inducible short hairpin RNA interference. Stem cells, 27, 776–82. doi: 10.1002/stem.5 .
- Zhang, F., and Lupski, J. R. (2015). Non-coding genetic variants in human disease. Human Molecular Genetics, 24, R102–R110. doi: 10.1093/hmg/ddv259 .
- Zwaka, T. P., and Thomson, J. A. (2003). Homologous recombination in human embryonic stem cells. Nature biotechnology, 21, 319–21. doi: 10.1038/nbt788 .